

Precision medicine is an emerging approach for the diagnosis, treatment and prognosis of genetic diseases that enables clinicians to more accurately predict which treatment strategy will be optimal in a patient. The aim of Precision Medicine in Oncology is to integrate clinical, histological, and molecular data in order to obtain a deeper knowledge about the biology and genetics of an individual's tumour. Over the last few years, the implementation of new NGS (Next Generation Sequencing) technologies into clinical practice has been essential. There is a wide variety of NGS techniques that can be used in this context. The correct interpretation of molecular changes detected by these techniques is paramount for their appropriate use. In this review, a discussion is presented on the main NGS sequencing technologies that can be used to improve the diagnosis, prognosis, and treatment of oncology patients.
El concepto de medicina de precisión ha cobrado especial relevancia en los últimos tiempos debido a la creciente necesidad de desarrollar estrategias personalizadas para el diagnóstico, el tratamiento y el seguimiento de diversas enfermedades de origen genético. La medicina de precisión en Oncología, a través de la integración de los datos clínicos, anatomopatológicos y moleculares, permite obtener un conocimiento más profundo del perfil biológico tumoral de cada paciente. En este contexto ha sido fundamental la implementación de las nuevas tecnologías de secuenciación next generation sequencing (NGS) en la práctica clínica. Existe un gran abanico de técnicas de secuenciación NGS que pueden ser utilizadas en función de la aplicación que se les quiera dar. La correcta interpretación de los cambios moleculares detectados mediante estas técnicas es clave para su adecuado uso en la práctica clínica. Esta revisión tiene como objetivo repasar las diferentes tecnologías de secuenciación que se utilizan actualmente en medicina de precisión para mejorar el diagnóstico, el pronóstico y el tratamiento de pacientes oncológicos.
The Human Genome Project, along with the development of high-throughput omic technologies such as next generation sequencing (NGS), has allowed rapid advances in our knowledge of disease-causing genetic alterations. The use of genetic data in clinical practice has led to a world-wide medical and scientific revolution, giving rise to a new model of health care delivery known as precision medicine or personalised medicine. There is abundant information on these emerging genomic technologies and their application to the diagnosis, prognosis and treatment of certain diseases. The aim of this review is to offer a clear and simple summary of the current situation of precision medicine in paediatric oncology, the most-recently developed sequencing technologies used in this field, and the suitability of this approach from the perspective of our experience in the Genomics and Paediatric Oncology units of the Instituto de Investigación Sanitaria and Hospital La Fe in Valencia.
What is precision medicine?The concept of precision medicine is not new. In the past few decades, one of the pillars of oncology has been the application of tumour information at the gene, protein and environment levels in diagnosis and treatment.1 The process of integrating clinical, histological and molecular data for the purpose of choosing the optimal or most suitable treatment for the biological profile of the tumour is what constitutes precision medicine.2
Traditionally, the molecular diagnosis of cancer based on the detection of single-nucleotide changes or small insertions or deletions in one or several genes was made by means of the Sanger sequencing method. Nowadays, due to the increasing number of genes that are known to be involved in cancer, the use of this technique is time-consuming and costly. Furthermore, it requires large amounts of tumour DNA, which may be difficult to obtain in some cases, and cannot detect DNA changes in subclonal populations. The development of NGS has revolutionised the approach to oncological diagnosis and provides more thorough and clinically applicable information on the molecular biology of tumours. Thanks to the development of these new technologies in combination with bioinformatic data analysis applications, the identification of pathogenic genomic alterations and of new genes associated with the development of specific diseases is now easier, faster and more cost-effective.
Genetics of childhood cancersThe annual incidence of paediatric cancer in developed countries is 140–160 de novo cases per million children aged 0–14 years. Although the cure rate is higher than in adults, paediatric cancer is the leading cause of death by disease in children aged more than 1 year.3 In Spain, approximately 1000 cases of childhood cancer are diagnosed each year, an incidence that rises to 1500 for age up to 18 years.4 Despite significant increases in childhood cancer survival in recent decades, the prognosis of some tumour groups, such as high-grade glioma, brainstem tumours, medulloblastoma, and metastatic sarcomas and neuroblastomas, continues to be poor. Furthermore, survival is less than 20% in cases with metastatic relapse or progression, regardless of the used treatments.3
Given the heterogeneous characteristics and genetic complexity of many tumours,5 there is great variability in their response to treatment based on the altered molecular pathway. This is why an understanding of the molecular profile of the tumour is key to choose the most suitable treatment. In recent years, there have been significant advances in the molecular characterisation of different types of cancer that are invaluable tools for precision medicine.6
In addition to identify the genetic alterations that cause tumours, it is important to determine whether they are only found in the tumour (somatic mutation) or are constitutional in the patient and thus present in the blood (germline mutation). While only 5–10% of cancers are hereditary,7 there are several types of germline cancer caused by known mutations. These cases are diagnosed differently and carry a different prognosis, and require genetic counselling since they not only increase the risk of carriers to develop cancer, but may also affect other members of the family and be passed on to the offspring. Recent studies have shown that approximately half of the patients with germline cancers do not have a relevant family history of cancer.8
A new biopsy for molecular analysis should be performed for every tumour relapse, as the pattern of genetic alterations may change from the one that was identified at the time of diagnosis. A study in patients with neuroblastoma showed that the mutation burden increases significantly in relapses compared to the time of diagnosis, so that molecular testing of relapsing tumours can be very useful to identify potential treatment targets on a case-to-case basis.9
At the molecular level, paediatric cancers differ from adult cancers in both the type and frequency of genetic defects.10 Most paediatric tumours form in growing tissues during early organ development.
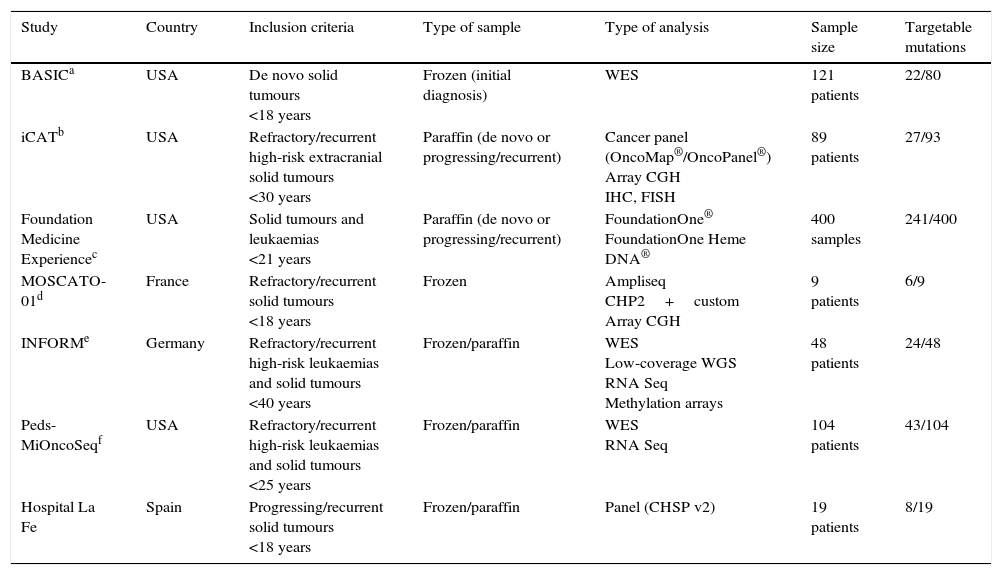
Several international initiatives have been launched with the purpose of elucidating cancer biology. The Cancer Genome Atlas is an ambitious project funded by the National Institute of Health of the United States whose objective is to make an exhaustive characterisation of each type of somatic cancer, in order to integrate all the information to establish the causes of the disease. The International Cancer Genome Consortium is an international project that is sequencing 50 tumour types. In the field of childhood cancer, various projects have been recently launched in the United States and Europe with the purpose of developing nationwide genetic screening programmes using commercially available gene panels for known mutations or whole exome and RNA sequencing (Table 1).11–13
Nationwide tumour molecular screening projects in the United States and Europe.
| Study | Country | Inclusion criteria | Type of sample | Type of analysis | Sample size | Targetable mutations |
|---|---|---|---|---|---|---|
| BASICa | USA | De novo solid tumours <18 years | Frozen (initial diagnosis) | WES | 121 patients | 22/80 |
| iCATb | USA | Refractory/recurrent high-risk extracranial solid tumours <30 years | Paraffin (de novo or progressing/recurrent) | Cancer panel (OncoMap®/OncoPanel®) Array CGH IHC, FISH | 89 patients | 27/93 |
| Foundation Medicine Experiencec | USA | Solid tumours and leukaemias <21 years | Paraffin (de novo or progressing/recurrent) | FoundationOne® FoundationOne Heme DNA® | 400 samples | 241/400 |
| MOSCATO-01d | France | Refractory/recurrent solid tumours <18 years | Frozen | Ampliseq CHP2+custom Array CGH | 9 patients | 6/9 |
| INFORMe | Germany | Refractory/recurrent high-risk leukaemias and solid tumours <40 years | Frozen/paraffin | WES Low-coverage WGS RNA Seq Methylation arrays | 48 patients | 24/48 |
| Peds-MiOncoSeqf | USA | Refractory/recurrent high-risk leukaemias and solid tumours <25 years | Frozen/paraffin | WES RNA Seq | 104 patients | 43/104 |
| Hospital La Fe | Spain | Progressing/recurrent solid tumours <18 years | Frozen/paraffin | Panel (CHSP v2) | 19 patients | 8/19 |
CGH, comparative genomic hybridisation; FISH, fluorescence in situ hybridisation; IHC, immunohistochemistry; WES, whole exome sequencing; WGS, whole genome sequencing.
In the past decade, we have progressed from sequencing a maximum of ninety-six 800 base pair sequences with first generation systems (Sanger method) to sequencing millions of DNA fragments with second generation systems (NGS). This novel technology is based on the amplification of DNA immobilised on a solid surface and the parallel reading of millions of sequences. The simultaneous sequencing of immobilised DNA reduces the amount of reactants needed as well as sequencing time, greatly reducing the cost per sequenced nucleotide. The vast amount of data generated by this technology poses a challenge to bioinformaticians, requiring the development of specific and user-friendly data analysis applications.
In addition to the detection of point mutations, mass sequencing allows the detection of insertions, deletions, gene copy number changes and translocations. Another important aspect of the use of NGS in the study of cancer is the detection of somatic variants in tumour cell subpopulations that, as such, are present in low proportions in tumour samples. These subclonal mutations, which are undetectable by Sanger sequencing, are sometimes responsible for the recurrence or resistance to treatment of some tumours.18
We now proceed to describe the main NGS methods used in cancer diagnosis, along with their advantages and limitations:
- –
Gene panel sequencing: panels contain primers or probes for a known group of genes and allow targeted sequencing for a specific disease. There are many commercially available panels, although they can also be made a la carte. They can be used to sequence known mutation regions (“hot spots”) and full genes, and allow the detection of gene copy number changes and translocations.
Advantage: their design is optimised to sequence genes of interest with a high depth of coverage. This allows the detection of low-frequency variants and a quick and reliable analysis.
Limitation: at present, commercial cancer panels are designed for testing in adults, and do not include regions that are relevant in childhood cancers. Furthermore, since they target known regions, they do not allow the detection of new genes potentially involved in cancer.
- –
Whole exome sequencing (WES): the exome is the part of the genome corresponding to the coding regions (exons) that can be expressed and lead to the production of proteins. It comprehends approximately 1.5% of the genome and is its most important functional part. From a technical standpoint, there are different possible approaches to sequencing the exome, such as the previous amplification of exons by polymerase chain reaction or their capture by specific probes.
Advantage: allows the identification of genes and variants potentially involved in disease that have not yet been described.
Limitations: due to the greater number of regions to be sequenced, it requires more readings than gene panel sequencing, and it is more feasible from an economic standpoint if a lower coverage depth is used, which precludes the detection of subclonal mutations. Furthermore, its analysis and interpretation are more complex due to the large number of variants that are detected (approximately 40,000), so in cancer patients it is recommended that paired-end sequencing of tumour samples and matched blood is performed to determine which of the variants are somatic.
- –
Whole genome sequencing (WGS): comprehends the entire genome of the individual, including chromosomal and mitochondrial DNA.
Advantages: allows the identification of noncoding variants that may be associated to the disease. It is mainly used in research.
Limitations: it has a high cost and the analysis is very complex, as noncoding regions of the genome are less conserved and present a higher number of variants. Furthermore, it requires high-throughput sequencing systems that are not available in the laboratories of most research facilities and hospitals in Spain. Consequently, it is less feasible for the purposes of diagnosis in everyday clinical practice.
- –
Whole transcriptome sequencing (WTS) or RNA sequencing: it can provide quantitative data on the genes expressed at a given time.
Advantages: it allows the analysis of RNA transcripts, transcript isoforms, post-transcriptional modifications, gene fusion, mutations and changes in gene expression. It can be used to sequence different RNA populations (total RNA, small RNA, transfer RNA and ribosomal RNA).
Limitations: the results obtained are tissue-specific and time-dependent, and may vary with sequencing depth. Their analysis and interpretation is very complex, and it is recommended that expression in healthy tissue be used for control.
The correct interpretation of gene variants or detected mutations is key in precision medicine. Variants are detected by identifying differences in the DNA sequence of an individual compared to a reference sequence. However, the detection of variants in itself is not sufficient, as variants need to be interpreted by specialists to accurately determine their molecular and clinical implications and decide on the best treatment approach.19
Databases of variants detected in the healthy population (polymorphisms) and of pathogenic variants are available for the interpretation of variants. In the case of unknown variants, bioinformatic applications are used to predict pathogenicity in silico. Various parameters are used to identify pathogenic variants and exclude polymorphisms:
- –
Minor allele frequency (MAF) in the population: frequency with which the least common allele appears in a specific population. A high MAF is associated with population polymorphisms.
- –
Coverage: number of times a nucleotide is sequenced. If the coverage of the variant is lower than expected based on tumour cellularity, it could be a technical artefact or a cell subclone with a different genomic profile. Artefacts tend to recur in different samples on the same sequencing lane and can be identified by means of a sequence viewer, such as IGV.
- –
Variant location: a variant may be exonic (which may lead to an amino acid change or be a synonymous mutation) or intronic (may or may not affect consensus splice sites leading to errors in protein coding).
In addition to these parameters, we would like to underscore the importance of recording every variant found in the sequencing of different samples with a single technique in an in-house laboratory database. This allows the easy detection of errors intrinsic to the technology (which recur in all samples) and frequent population polymorphisms, which facilitate the analysis and interpretation of data to a great extent.
It is important to establish which category variants fit into: benign, likely benign, pathogenic, likely pathogenic or of uncertain clinical significance. A variant is benign when its MAF in the general population is greater than 2% or when it is categorised as such in the consulted databases (such as COSMIC [http://cancer.sanger.ac.uk/cosmic] or ClinVar [http://www.ncbi.nlm.nih.gov/clinvar/]). In the field of childhood cancer, there is the limitation that most variants are not described in existing databases, which calls for the use of in silico prediction tools that estimate the pathogenicity of mutations based on various aspects, such as allele frequencies in variant databases (1000G, http://www.1000genomes.org/, ExAC http://exac.broadinstitute.org/), protein structure (SIFT [http://sift.jcvi.org/] and Polyphen2 [http://genetics.bwh.harvard.edu/pph2/]) or splicing site defects (NNSplice [http://www.fruitfly.org/seq_tools/splice.html]; HSF [http://www.umd.be/HSF/], NetGene 2 [http://www.cbs.dtu.dk/services/NetGene2/] and SpliceView [http://bioinfo4.itb.cnr.it/∼webgene/wwwspliceview_ex.html]). If the variant is not described in the consulted databases and several in silico prediction tools suggest that it is a benign mutation, it can be interpreted as a variant of uncertain clinical significance/likely benign. A variant is considered to be of uncertain clinical significance when it is not described in the consulted databases but could have an effect on gene function. The analysis of these variants involves the use of the aforementioned prediction tools and the estimation of a pathogenicity level by integrating other data contributed by pathologists, clinicians and bioinformaticians. A variant is considered pathogenic if different studies support its pathogenicity, and probably pathogenic when it is described as such in the literature or in the consulted databases, or when it has not been described but several prediction tools suggest that it is a pathogenic mutation.
Application of precision medicine to health care delivery. Opportunities for treatmentIn the past decade, most trials of molecularly targeted agents for the treatment of childhood cancers have been performed in unselected patients.20 At present, few molecularly targeted therapies are available in the field of paediatric oncology. Those that are most widely used consist of drugs originally developed for other diseases in adults, such as ALK inhibitors, used in non-small cell lung carcinoma with ALK gene rearrangement, which have also proven effective in the treatment of anaplastic large-cell lymphoma and inflammatory myofibroblastic tumour (with ALK rearrangement) and neuroblastomas (with a mutation in ALK),21 and BRAF inhibitors, used in melanoma, which are also effective in paediatric cases of BRAF V600E-mutated glioma.22,23 The molecular analysis of ALK and BRAF in tumour tissue samples is part of the standard diagnostic protocol in most paediatric oncology clinics. However, the consecutive analysis of biomarkers is not compatible with clinical practice due to the limited availability of tumour material (which is usually obtained by a core needle biopsy at the time of diagnosis), the time required for each test, and the overall cost of the process.24
Our own experience, which is consistent with the data reported by other institutions in Spain and abroad, shows that it is possible to incorporate NGS to the clinical management of childhood cancer patients.11–13 In November 2014, the Genomic Unit of the Instituto de Investigación Sanitaria La Fe (La Fe Public Health Research Institute) and the Paediatric Oncology Unit of the Hospital la Fe of Valencia established a close cooperation framework for the practice of precision medicine in paediatric patients with progressing or recurrent cancer.
Most of the samples submitted to the Genomic Unit are analysed using NGS panels. The findings are discussed every fortnight by a precision medicine committee formed by pathologists, molecular biologists, geneticists and paediatric oncologists. This team discusses the pathogenicity of the detected variants, whether these variants open up new therapeutic options, and whether there are ongoing clinical trials that would fit specific patients. Achieving this rapid laboratory-to-clinic translation with real benefits for the patient is the greatest challenge of paediatric oncology at the moment.
Applying the precision medicine protocol of our hospital, we evaluated 19 patients with progressing or recurrent solid tumours by means of NGS using a commercial hotspot mutation panel (CHSP v2; Thermofisher Scientific; Uppsala, Sweden). The median age at the time of inclusion in the study was 9.2 years (range, 3.6–16.4 years) and most patients had extracranial solid tumours (16), specifically neuroblastomas (7) and sarcomas (7).
The analysed samples contained a minimum of 50% tumour cells. We found mutations that were potential therapeutic targets in eight of the nineteen patients. A mean of nineteen days elapsed between the time the biopsy or surgery was performed and the time that the molecular oncology committee made the corresponding recommendation.
Three of the patients under study were treated with compassionate-use agents based on the genetic profile of their tumours. One of them received a diagnosis of Cowden syndrome based on genetic testing of a hamartomatous skin lesion that detected a de novo germline mutation (confirmed by testing of peripheral blood). This patient has been treated with everolimus and remained in partial remission after a year and a half of treatment. The second patient had a rapidly-progressing inflammatory myofibroblastic tumour that was resistant to ALK inhibitors and was treated with an mTOR inhibitor (temsirolimus) combined with irinotecan and temozolomide; the initial response was favourable, but the tumour started progressing after two months of treatment. The third patient had a neuroblastoma with a mutation in the ALK gene that progressed through several lines of treatment, and has recently enrolled in a phase I clinical trial for an ALK inhibitor.
At present, thanks to the rapid development of high-throughput technologies and supporting bioinformatic programmes, we can analyse tumours at the molecular level to an unprecedented degree. We expect that this ability to study biological phenomena at the omic level will continue to lead to significant advances in precision medicine and in the identification of new, potentially treatable mutations and less frequent genetic alterations for which targeted therapies are already available, improving the prognosis of our patients.
Conflict of interestThe authors have no conflict of interest to declare.
Please cite this article as: Calabria I, Pedrola L, Berlanga P, Aparisi MJ, Sánchez-Izquierdo D, Cañete A, et al. El nuevo reto en oncología: la secuenciación NGS y su aplicación a la medicina de precisión. An Pediatr (Barc). 2016;85:273.

