

Los cilios son organelas conservadas evolutivamente, presentes en la mayoría de las células polarizadas del organismo. El cilio emerge del cuerpo basal, extendiéndose desde la superficie celular al espacio extracelular constituyendo una estructura microtubular proteica o axonema. La posición del cilio en la superficie celular facilita su función como sensor y vehículo transmisor de información entre la célula y el espacio extracelular1–4.
Presentamos los casos de 4 pacientes, fenotípicamente diferentes que comparten un denominador común, la disfunción ciliar:
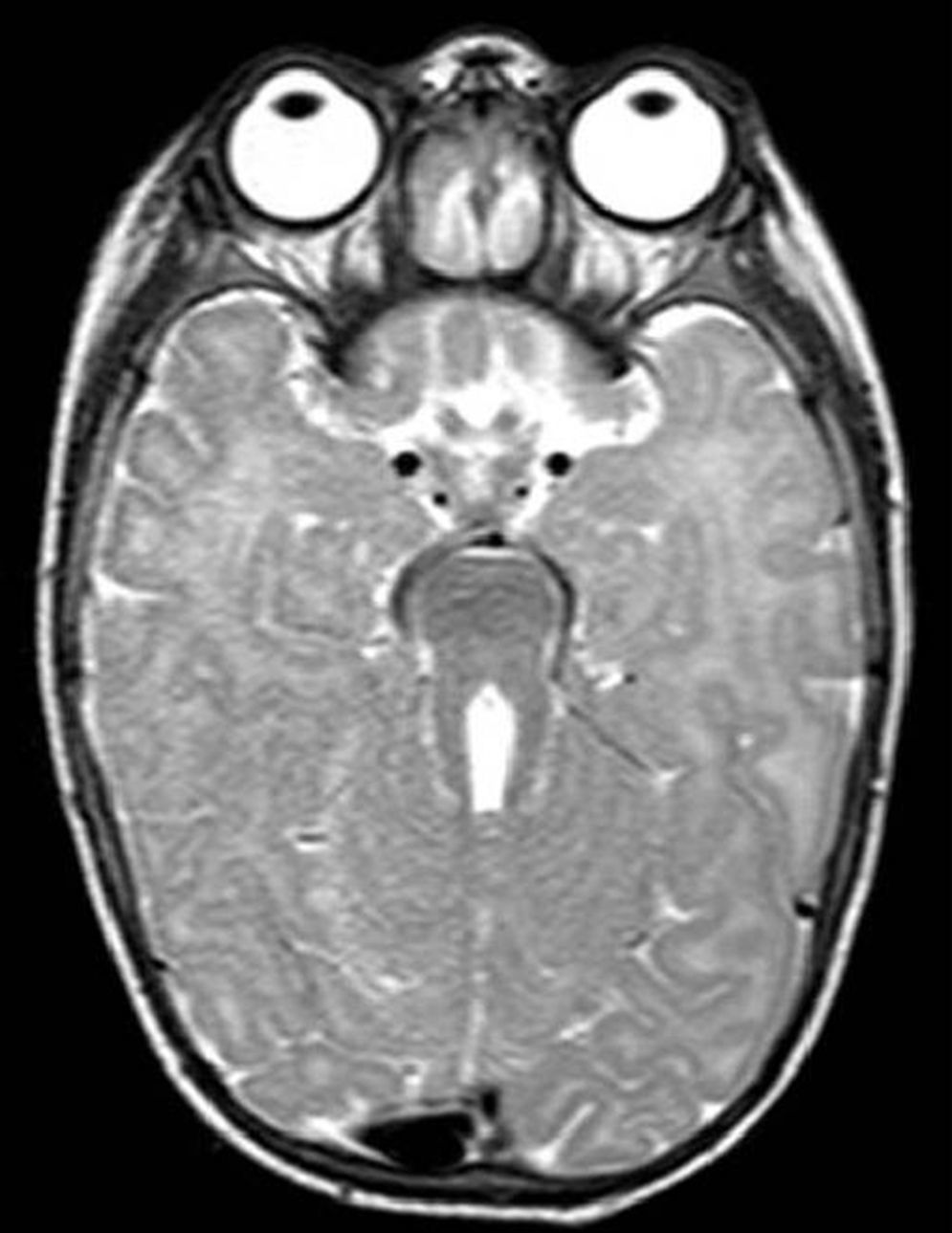
El primer caso es una lactante mujer de 6 meses con retraso psicomotor e hipotonía axial, nistagmo vertical y movimientos oculares erráticos. Prenatalmente se sospechó hipoplasia del vermis cerebeloso. En la RM cerebral posnatal se objetivó ausencia completa de vermis cerebeloso con alteración de pedúnculos cerebelosos y cuarto ventrículo con morfología «signo del molar» (fig. 1). El síndrome de Joubert (OMIM 213300) se confirmó al detectar la mutación c.2168G>A (pArg723Gln) en el gen AHI1 en homocigosis. Se caracteriza por malformación congénita de tronco cerebral y agenesia o hipoplasia vermis cerebeloso, que causa hipotonía y ataxia, retraso desarrollo motor, nistagmo y patrón respiratorio neonatal de taquipnea/disnea. El déficit intelectual es variable. Puede asociar distrofia retiniana, nefronoptisis y polidactilia, presentes en otras ciliopatías. Su herencia es autosómica recesiva, identificando mutaciones en distintos genes: AHI1, NPHP1, CEP290, TMEM67, RPGRIP1L, ARL13B y CC2D2A5. El síndrome de Bardet-Biedl, Meckel-Gruber, la amaurosis congénita de Leber y la nefronoptisis comparten mutaciones en gen CEP290, demostrando la superposición genética y clínica en los síndromes asociados a disfunción del cilio primario6.

El segundo paciente se trata de un recién nacido que inicia dificultad respiratoria con necesidad de oxigenoterapia, sin antecedentes perinatales de interés. Los padres son consanguíneos y su hermano anterior ingresó a los 12 días de vida por dificultad respiratoria con atelectasia lobar. Radiológicamente presenta atelectasia en LSI y situs inversus. Se sospecha discinesia ciliar primaria (OMIM 244400), ciliopatía motora que incluye el síndrome de Kartagener, generalmente autosómico recesiva. Se describen mutaciones en 11 genes: DNAI1, DNAH5, DNAH11, DNAI2, KTU, TXNDC3, LRRC50, RSPH9, RSPH4A, CCDC40 y CCDC39. La disfunción de las células ciliadas conduce a infecciones crónicas respiratorias. También afecta al flagelo del espermatozoide y a los cilios de la trompa de Falopio3,7. El establecimiento del eje de lateralidad izquierda-derecha embrionario es un mecanismo dependiente del cilio. La ineficacia de estos cilios nodales conlleva que la asimetría de los órganos internos se disponga al azar, por lo que el 50% presenta situs inversus totalis1,7.
El tercer paciente es un recién nacido con antecedentes prenatales de hidronefrosis derecha. Presenta polidactilia postaxial en ambas manos y leucocoria en el ojo izquierdo. Ecográficamente riñón derecho de 50mm con hidronefrosis grado III-IV, dilatación de la pelvis renal (30mm) y de todo el uréter hasta nivel distal, con prominente ureterocele. Precisa descompresión del ureterocele e intervención precoz de la catarata. El cuadro clínico es característico del síndrome de Bardet-Biedl (OMIM 209900), una ciliopatía sensorial recesiva con gran variabilidad clínica: obesidad, retinopatía pigmentaria, polidactilia postaxial, poliquistosis renal, hipogonadismo, retraso mental, diabetes mellitus y cardiopatía congénita. El amplio espectro clínico viene determinado por su heterogenicidad genética, describiéndose mutaciones en 12 genes (BBS1 hasta BBS12)2,3. El diagnóstico diferencial incluye el síndrome de Alström con el que comparte obesidad, diabetes mellitus, nefronoptisis y retinitis pigmentaria.
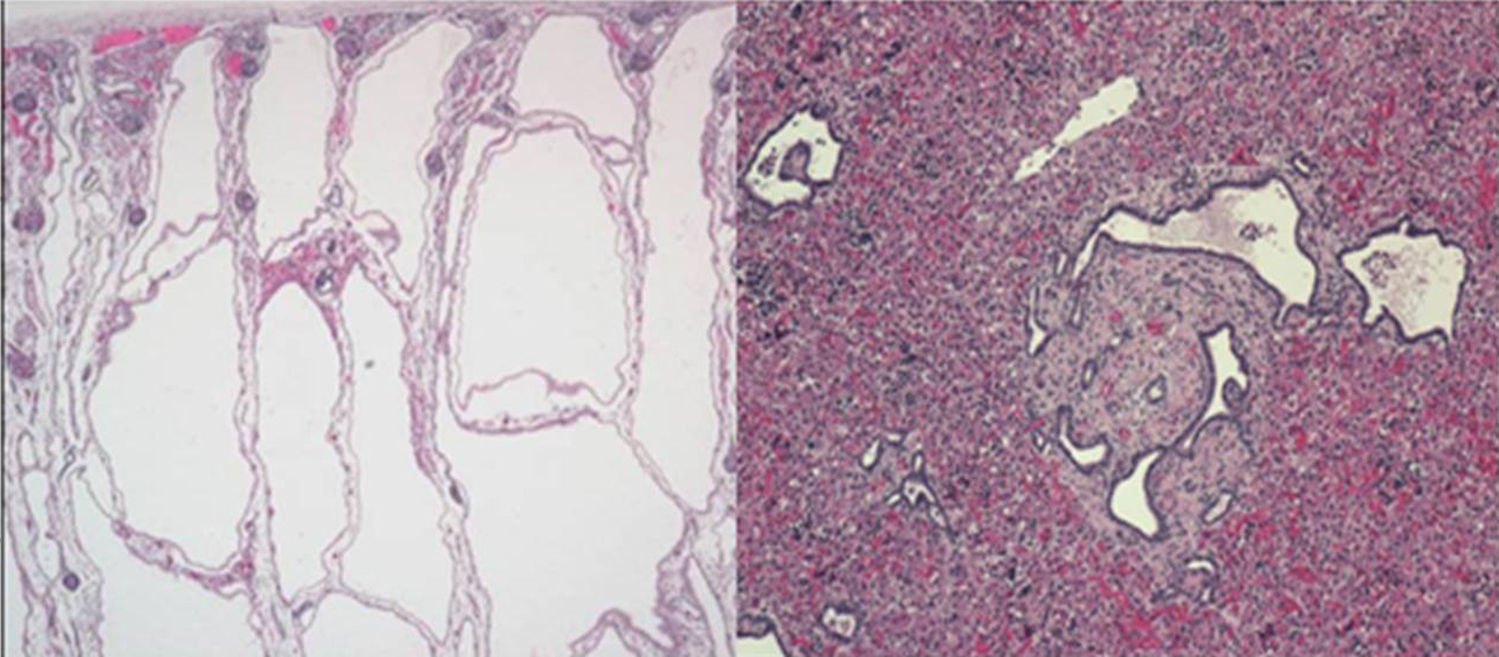
El último caso clínico se trata de un recién nacido pretérmino con oligoamnios y nefromegalia bilateral detectada anteparto. Antecedente paterno de hipertensión arterial e hipertransaminemia. Se realiza cesárea urgente, desarrollando dificultad respiratoria grave. Presenta tórax pequeño, en campana, con abdomen distendido, palpándose masas en ambos flancos. Radiológicamente neumotórax que no responde a drenaje e hipoplasia pulmonar. Evoluciona con anuria e hipotensión arterial. La ecografía abdominal muestra microquistes en ambos riñones e hígado, compatibles con enfermedad poliquística renal, autosómica recesiva. Éxitus a las 20h de vida. La necropsia confirma el diagnóstico de poliquistosis renal (OMIM 263200) (fig. 2). Se trata de la ciliopatía más frecuente en la primera infancia. Caracterizada por la dilatación de los túbulos colectores renales y progresiva degeneración quística renal y fibrosis hepática con mortalidad temprana. Se debe a la mutación en el gen PKHD1 que codifica la proteína poliductina encargada de la diferenciación de las células de revestimiento de los túbulos colectores renales2,3.

Las ciliopatías comprenden un grupo de entidades clínicas genéticamente heterogéneas debido a la complejidad molecular del axonema ciliar. Suponen un reto para la investigación por su participación en diversas enfermedades y vías de señalización, estando involucradas en la patogenia de la obesidad, la diabetes y la oncogénesis8–10.

