

El síndrome de Cornelia de Lange (SCdL) es una enfermedad rara congénita del desarrollo con afectación multisistémica. Las manifestaciones clínicas son muy variables, pero se distingue entre un fenotipo clásico, caracterizado por unos rasgos craneofaciales distintivos, retraso del crecimiento pre y posnatal, defectos por reducción de las extremidades, hirsutismo y discapacidad intelectual, y un fenotipo no clásico, generalmente más leve y más difícil de diagnosticar. Además, las características clínicas se superponen con las de otros trastornos del neurodesarrollo, por lo que la utilización de criterios clínicos consensuados y de herramientas de inteligencia artificial puede ser útil para confirmar el diagnóstico.
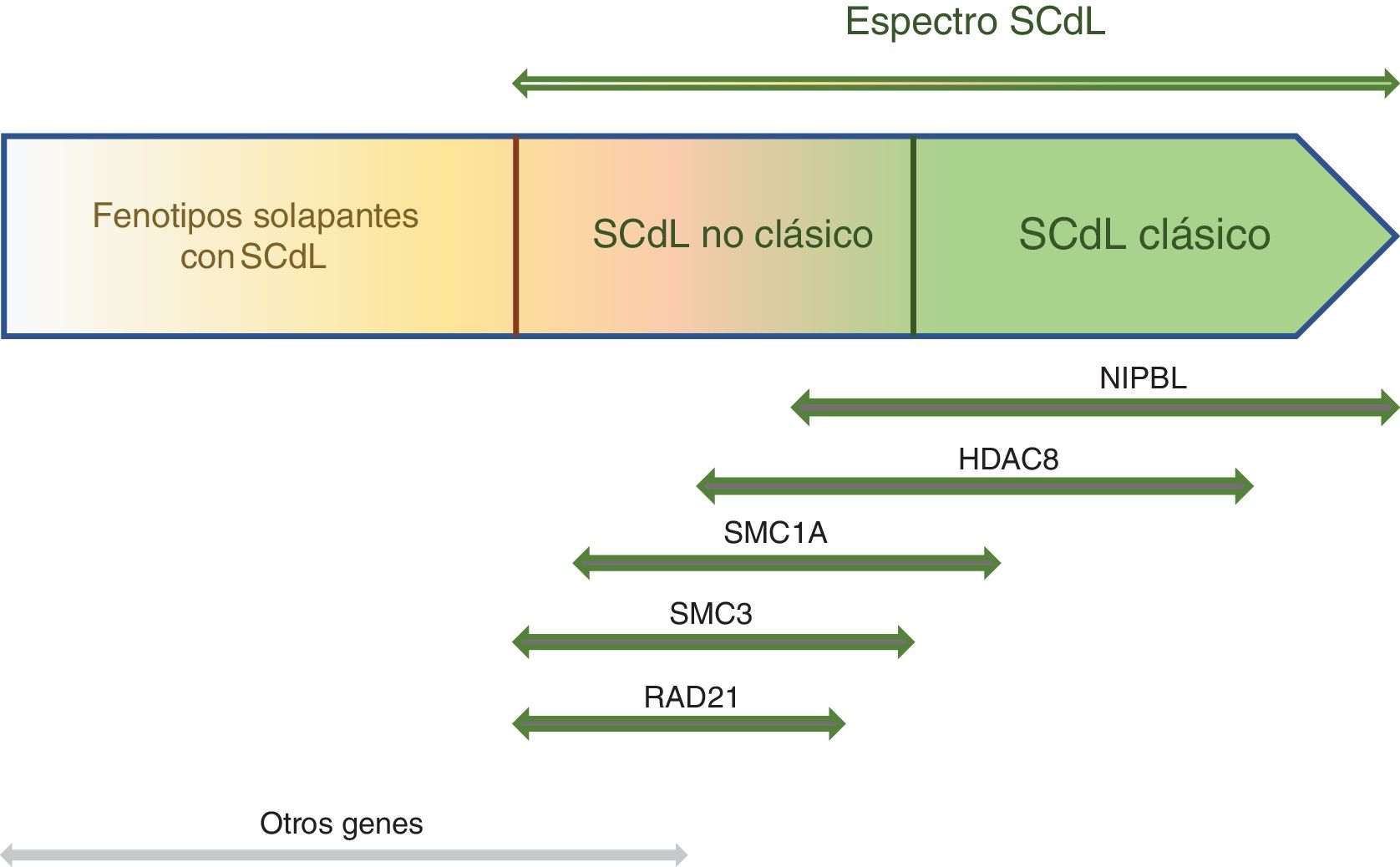
En más del 60% de los pacientes se han identificado variantes patogénicas en el gen NIPBL, que codifica una proteína relacionada con el complejo de la cohesina, y en otro 15% en 4 genes también asociados a este complejo: SMC1A, SMC3, RAD21 y HDAC8. Los progresos en las técnicas de secuenciación masiva han permitido describir otros genes relacionados (BRD4, ANKRD11 y MAU2), pero la ausencia de diagnóstico molecular en el 15% de los casos y la gran heterogeneidad clínica del síndrome sugieren la existencia de otros genes y mecanismos relacionados.
Aunque no haya un tratamiento curativo, sí hay tratamientos sintomáticos/paliativos que deben ser conocidos por el pediatra. La principal complicación médica en el SCdL clásico es el reflujo gastroesofágico, que debe ser tratado de forma precoz y contundente, ya que es una de las causas más frecuentes de fallecimiento en estos pacientes.
Cornelia de Lange syndrome (CdLS) is a rare congenital developmental disorder with multisystemic involvement. The clinical presentation is highly variable, but the classic phenotype, characterized by distinctive craniofacial features, pre- and postnatal growth retardation, extremity reduction defects, hirsutism and intellectual disability can be distinguished from the nonclassic phenotype, which is generally milder and more difficult to diagnose. In addition, the clinical features overlap with those of other neurodevelopmental disorders, so the use of consensus clinical criteria and artificial intelligence tools may be helpful in confirming the diagnosis.
Pathogenic variants in NIPBL, which encodes a protein related to the cohesin complex, have been identified in more than 60% of patients, and pathogenic variants in other genes related to this complex in another 15%: SMC1A, SMC3, RAD21, and HDAC8. Technical advances in large-scale sequencing have allowed the description of additional genes (BRD4, ANKRD11, MAU2), but the lack of molecular diagnosis in 15% of individuals and the substantial clinical heterogeneity of the syndrome suggest that other genes and mechanisms may be involved.
Although there is no curative treatment, there are symptomatic/palliative treatments that paediatricians should be aware of. The main medical complication in classic CdLS is gastro-oesophageal reflux, which should be treated early.
El síndrome de Cornelia de Lange (SCdL) es una enfermedad rara que debe su nombre a la pediatra holandesa Cornelia de Lange, quien en 1933 publicó los casos de dos niñas pequeñas sin parentesco familiar que tenían un fenotipo muy similar1 El cuadro clínico lo denominó typus degenerativus amstelodamensis, en honor a la ciudad de Ámsterdam, donde trabajaba. Previamente, en 1916, Brachmann había publicado un paciente con retraso de crecimiento, hirsutismo y monodactilia bilateral, que posteriormente fue considerado como el primer caso documentado de SCdL2, por lo que en algunas publicaciones el cuadro se denomina síndrome de Brachmann-de Lange.
Actualmente la prevalencia del SCdL se estima entre 1:10.000 y 1:30.000 individuos de la población general3. Estas cifras incluyen los casos con formas no clásicas de SCdL, que en décadas pasadas no se diagnosticaban al no disponerse de los estudios genéticos actuales.
Formas clínicasSíndrome Cornelia de Lange clásicoEl SCdL clásico, reconocible al nacimiento, se caracteriza por un fenotipo físico con rasgos craneofaciales característicos, retraso del crecimiento pre y posnatal, defectos de reducción de las extremidades (principalmente superiores), hirsutismo y retraso psicomotor/discapacidad intelectual. Son frecuentes las anomalías en diferentes sistemas: reflujo gastroesofágico (la complicación médica más frecuente e importante en pacientes con SCdL), hipoacusia neurosensorial, cardiopatía congénita, anomalías renales y genitales, entre otras3,4.
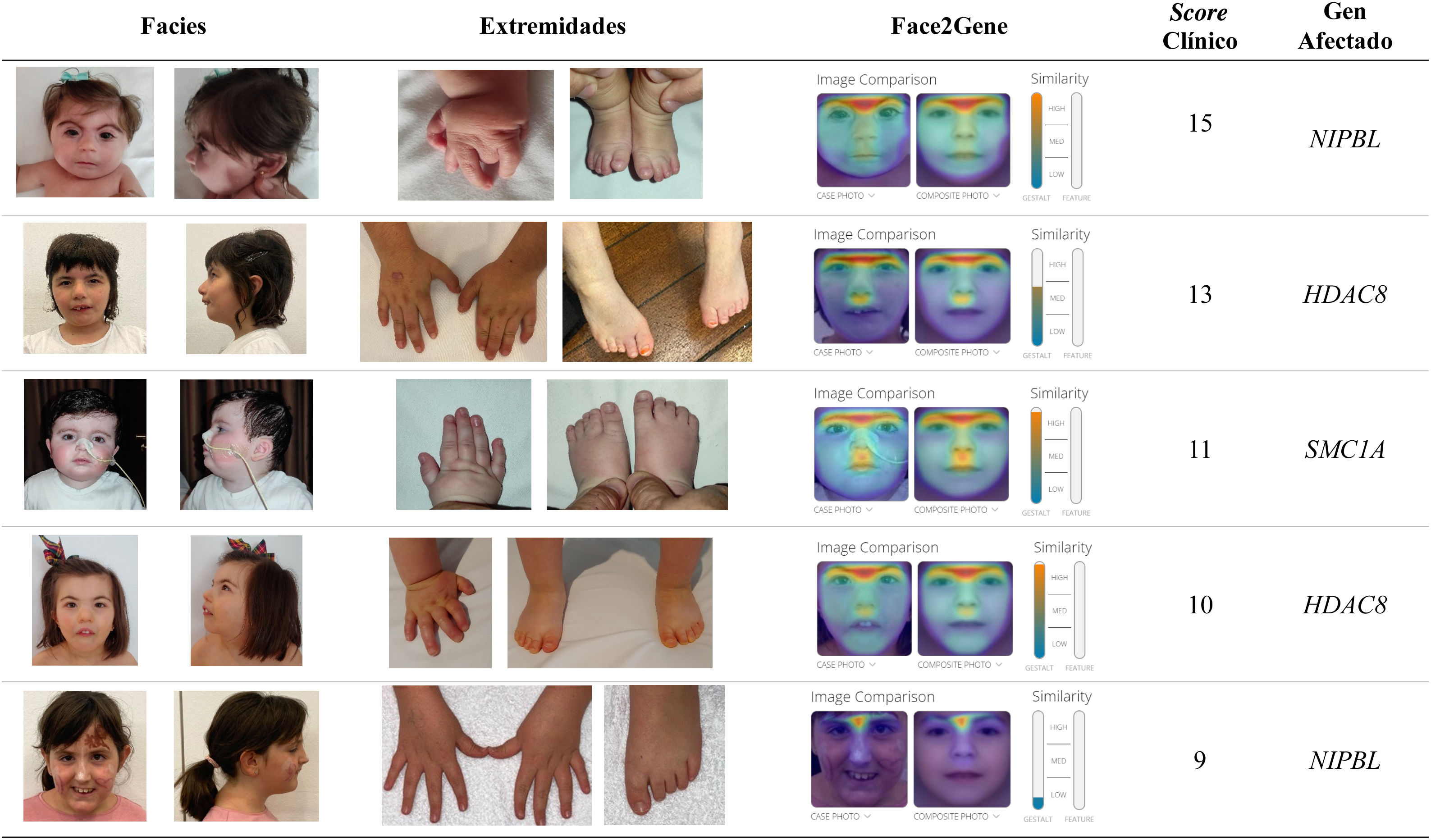
Los rasgos craneofaciales característicos incluyen microcefalia con braquicefalia, sinofria, cejas pobladas arqueadas, pestañas largas, nariz corta con puente nasal hundido y narinas antevertidas, filtro alargado con labio superior fino y retrognatia3,4 (fig. 1 y tabla 1).

Rasgos faciales y alteraciones en las extremidades en pacientes con SCdL y variantes patogénicas en diferentes genes causales. Se muestra también el análisis mediante el programa «Face2Gene» y los scores calculados según Kline et al3.
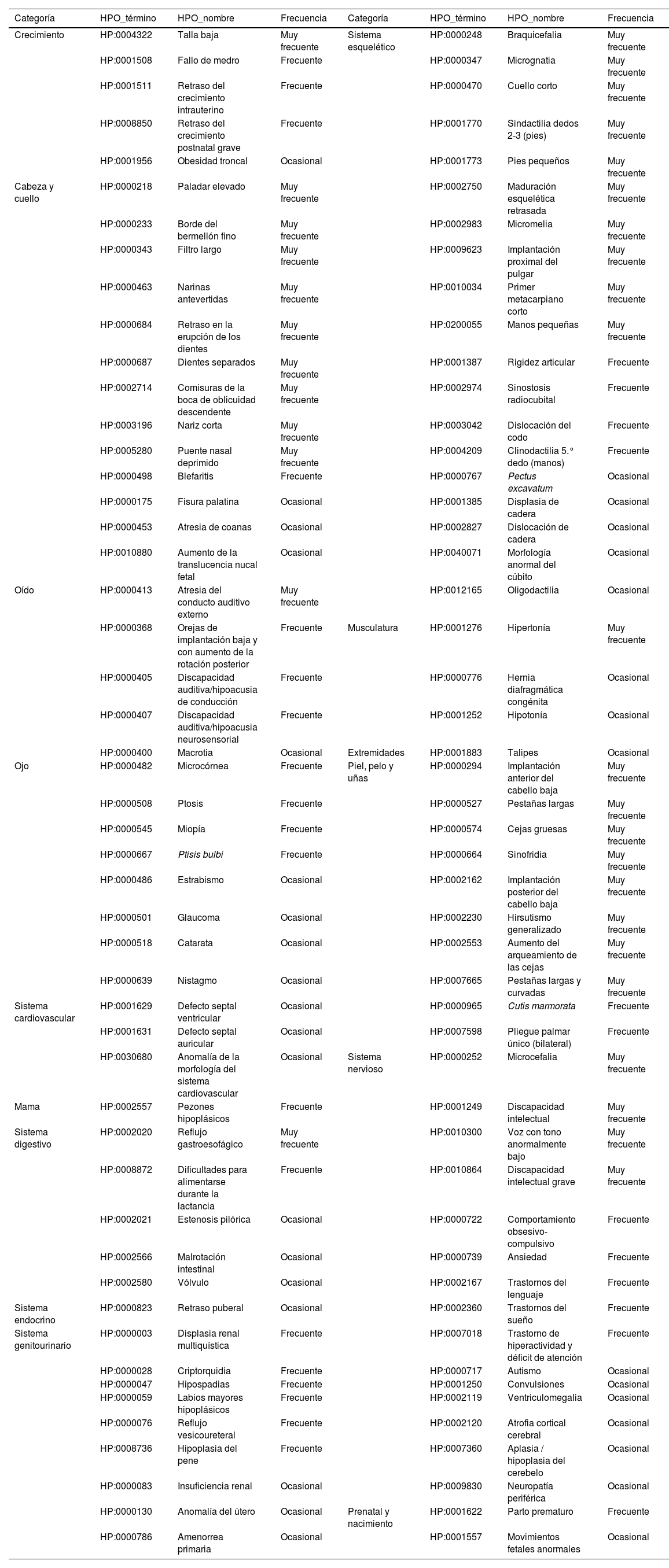
Espectro clínico del SCdL ordenado por categorías, hallazgos definidos y numerados según el sistema HPO (Human Phenotype Ontology), y frecuencia de aparición
| Categoría | HPO_término | HPO_nombre | Frecuencia | Categoría | HPO_término | HPO_nombre | Frecuencia |
|---|---|---|---|---|---|---|---|
| Crecimiento | HP:0004322 | Talla baja | Muy frecuente | Sistema esquelético | HP:0000248 | Braquicefalia | Muy frecuente |
| HP:0001508 | Fallo de medro | Frecuente | HP:0000347 | Micrognatia | Muy frecuente | ||
| HP:0001511 | Retraso del crecimiento intrauterino | Frecuente | HP:0000470 | Cuello corto | Muy frecuente | ||
| HP:0008850 | Retraso del crecimiento postnatal grave | Frecuente | HP:0001770 | Sindactilia dedos 2-3 (pies) | Muy frecuente | ||
| HP:0001956 | Obesidad troncal | Ocasional | HP:0001773 | Pies pequeños | Muy frecuente | ||
| Cabeza y cuello | HP:0000218 | Paladar elevado | Muy frecuente | HP:0002750 | Maduración esquelética retrasada | Muy frecuente | |
| HP:0000233 | Borde del bermellón fino | Muy frecuente | HP:0002983 | Micromelia | Muy frecuente | ||
| HP:0000343 | Filtro largo | Muy frecuente | HP:0009623 | Implantación proximal del pulgar | Muy frecuente | ||
| HP:0000463 | Narinas antevertidas | Muy frecuente | HP:0010034 | Primer metacarpiano corto | Muy frecuente | ||
| HP:0000684 | Retraso en la erupción de los dientes | Muy frecuente | HP:0200055 | Manos pequeñas | Muy frecuente | ||
| HP:0000687 | Dientes separados | Muy frecuente | HP:0001387 | Rigidez articular | Frecuente | ||
| HP:0002714 | Comisuras de la boca de oblicuidad descendente | Muy frecuente | HP:0002974 | Sinostosis radiocubital | Frecuente | ||
| HP:0003196 | Nariz corta | Muy frecuente | HP:0003042 | Dislocación del codo | Frecuente | ||
| HP:0005280 | Puente nasal deprimido | Muy frecuente | HP:0004209 | Clinodactilia 5.° dedo (manos) | Frecuente | ||
| HP:0000498 | Blefaritis | Frecuente | HP:0000767 | Pectus excavatum | Ocasional | ||
| HP:0000175 | Fisura palatina | Ocasional | HP:0001385 | Displasia de cadera | Ocasional | ||
| HP:0000453 | Atresia de coanas | Ocasional | HP:0002827 | Dislocación de cadera | Ocasional | ||
| HP:0010880 | Aumento de la translucencia nucal fetal | Ocasional | HP:0040071 | Morfología anormal del cúbito | Ocasional | ||
| Oído | HP:0000413 | Atresia del conducto auditivo externo | Muy frecuente | HP:0012165 | Oligodactilia | Ocasional | |
| HP:0000368 | Orejas de implantación baja y con aumento de la rotación posterior | Frecuente | Musculatura | HP:0001276 | Hipertonía | Muy frecuente | |
| HP:0000405 | Discapacidad auditiva/hipoacusia de conducción | Frecuente | HP:0000776 | Hernia diafragmática congénita | Ocasional | ||
| HP:0000407 | Discapacidad auditiva/hipoacusia neurosensorial | Frecuente | HP:0001252 | Hipotonía | Ocasional | ||
| HP:0000400 | Macrotia | Ocasional | Extremidades | HP:0001883 | Talipes | Ocasional | |
| Ojo | HP:0000482 | Microcórnea | Frecuente | Piel, pelo y uñas | HP:0000294 | Implantación anterior del cabello baja | Muy frecuente |
| HP:0000508 | Ptosis | Frecuente | HP:0000527 | Pestañas largas | Muy frecuente | ||
| HP:0000545 | Miopía | Frecuente | HP:0000574 | Cejas gruesas | Muy frecuente | ||
| HP:0000667 | Ptisis bulbi | Frecuente | HP:0000664 | Sinofridia | Muy frecuente | ||
| HP:0000486 | Estrabismo | Ocasional | HP:0002162 | Implantación posterior del cabello baja | Muy frecuente | ||
| HP:0000501 | Glaucoma | Ocasional | HP:0002230 | Hirsutismo generalizado | Muy frecuente | ||
| HP:0000518 | Catarata | Ocasional | HP:0002553 | Aumento del arqueamiento de las cejas | Muy frecuente | ||
| HP:0000639 | Nistagmo | Ocasional | HP:0007665 | Pestañas largas y curvadas | Muy frecuente | ||
| Sistema cardiovascular | HP:0001629 | Defecto septal ventricular | Ocasional | HP:0000965 | Cutis marmorata | Frecuente | |
| HP:0001631 | Defecto septal auricular | Ocasional | HP:0007598 | Pliegue palmar único (bilateral) | Frecuente | ||
| HP:0030680 | Anomalía de la morfología del sistema cardiovascular | Ocasional | Sistema nervioso | HP:0000252 | Microcefalia | Muy frecuente | |
| Mama | HP:0002557 | Pezones hipoplásicos | Frecuente | HP:0001249 | Discapacidad intelectual | Muy frecuente | |
| Sistema digestivo | HP:0002020 | Reflujo gastroesofágico | Muy frecuente | HP:0010300 | Voz con tono anormalmente bajo | Muy frecuente | |
| HP:0008872 | Dificultades para alimentarse durante la lactancia | Frecuente | HP:0010864 | Discapacidad intelectual grave | Muy frecuente | ||
| HP:0002021 | Estenosis pilórica | Ocasional | HP:0000722 | Comportamiento obsesivo-compulsivo | Frecuente | ||
| HP:0002566 | Malrotación intestinal | Ocasional | HP:0000739 | Ansiedad | Frecuente | ||
| HP:0002580 | Vólvulo | Ocasional | HP:0002167 | Trastornos del lenguaje | Frecuente | ||
| Sistema endocrino | HP:0000823 | Retraso puberal | Ocasional | HP:0002360 | Trastornos del sueño | Frecuente | |
| Sistema genitourinario | HP:0000003 | Displasia renal multiquística | Frecuente | HP:0007018 | Trastorno de hiperactividad y déficit de atención | Frecuente | |
| HP:0000028 | Criptorquidia | Frecuente | HP:0000717 | Autismo | Ocasional | ||
| HP:0000047 | Hipospadias | Frecuente | HP:0001250 | Convulsiones | Ocasional | ||
| HP:0000059 | Labios mayores hipoplásicos | Frecuente | HP:0002119 | Ventriculomegalia | Ocasional | ||
| HP:0000076 | Reflujo vesicoureteral | Frecuente | HP:0002120 | Atrofia cortical cerebral | Ocasional | ||
| HP:0008736 | Hipoplasia del pene | Frecuente | HP:0007360 | Aplasia / hipoplasia del cerebelo | Ocasional | ||
| HP:0000083 | Insuficiencia renal | Ocasional | HP:0009830 | Neuropatía periférica | Ocasional | ||
| HP:0000130 | Anomalía del útero | Ocasional | Prenatal y nacimiento | HP:0001622 | Parto prematuro | Frecuente | |
| HP:0000786 | Amenorrea primaria | Ocasional | HP:0001557 | Movimientos fetales anormales | Ocasional |
Frecuencia según Human Phenotype Ontology (HPO): https://hpo.jax.org/app/. Muy frecuente 99-80%; Frecuente 79-30%; Ocasional 29-5%.
El retraso del crecimiento es muy frecuente y es de inicio prenatal. Los pacientes presentan una microsomía generalizada y simétrica que da lugar a un bajo peso, una talla baja y una microcefalia que permanecen hasta la edad adulta5,6. Todo ello ha llevado al desarrollo de gráficas específicas para el síndrome7. Aunque los niveles de hormona de crecimiento suelen ser normales, algunos pacientes con valores bajos han sido tratados con esta hormona8,9.
El retraso psicomotor/discapacidad intelectual está presente en la mayoría de pacientes con SCdL clásico, siendo generalmente grave/profundo10. Se suele asociar ausencia o retraso del lenguaje y trastornos del espectro autista y del comportamiento. Desde el punto de vista neurológico, los pacientes pueden tener epilepsia, trastornos motores (movimientos estereotipados) y sensitivos (sensibilidad al dolor disminuida o intolerancia al frío o calor), hipotonía o problemas con el sueño. Recientemente se ha descrito también la afectación del sistema nervioso autónomo con neuropatía de fibra fina acompañada, a veces, de asimetrías en las respuestas simpáticas en los miembros inferiores11.
Las anomalías de extremidades se presentan en alrededor del 80% de los casos. Entre un 25 y un 30% tienen malformaciones graves, principalmente defectos de reducción en las extremidades superiores, como monodactilia u oligodactilia, mientras que el resto suelen tener manos y pies pequeños que pueden estar acompañados de malformaciones menores, como pulgares de implantación distal o clinodactilia. Estas anomalías suelen ser bilaterales y generalmente asimétricas12 (fig. 1, tabla 1).
El reflujo gastroesofágico (RGE) es el principal problema médico en el SCdL clásico, y afecta al 93,3% de los pacientes13,14. Debe ser investigado desde edades tempranas en todo paciente con problemas de alimentación, realizando una pHmetría/manometría o, en su defecto, estudio del tracto digestivo superior con contraste de bario. Si no se trata adecuadamente (ver apartado «Tratamiento y seguimiento») puede dar lugar a complicaciones como esofagitis, neumonitis/neumonía por aspiración y alteraciones del comportamiento (irritabilidad, autoagresividad, etc.), que muchas veces son tratadas ineficazmente con fármacos psicotropos.
Otras anomalías digestivas, mucho menos frecuentes, incluyen la malrotación intestinal (2-3% de casos), que debe tenerse siempre presente en pacientes con SCdL que acuden a urgencias por un cuadro de dolor abdominal agudo, la estenosis pilórica (7%) o la hernia diafragmática congénita15.
En el área sensorial, la complicación más frecuente es la hipoacusia neurosensorial, presente en el 80% de los pacientes, de los cuales casi la mitad tienen una hipoacusia bilateral profunda16. En el área oftalmológica son frecuentes la ptosis palpebral, generalmente bilateral, y la miopía (>50% casos) y el nistagmo (40%). Menos frecuentes son la estenosis del conducto lacrimal, el estrabismo, la microcórnea, el glaucoma o el coloboma del nervio óptico17.
En el sistema cardiovascular, alrededor de un tercio de pacientes tienen una cardiopatía congénita, siendo las anomalías más frecuentes los defectos del tabique ventricular o auricular, estenosis pulmonar, tetralogía de Fallot, síndrome del corazón izquierdo hipoplásico y válvula aórtica bicúspide18.
Un estudio reciente ha demostrado que algunos individuos pueden presentar miocardiopatía temprana, que puede ser detectada mediante la técnica de speckle tracking incluso antes de la aparición de síntomas clínicos y de la alteración de otros parámetros ecocardiográficos o analíticos19.
En el sistema genitourinario son frecuentes la criptorquidia en varones (75%), el útero bicorne en mujeres (25%) y genitales hipoplásicos en ambos sexos (50%). Las anomalías renales son poco frecuentes, siendo el reflujo vesiculoureteral la más prevalente20 (10%) (tabla 1).
Se ha descrito también una cierta desregulación del sistema endocrino, con niveles alterados de prolactina y pubertad retrasada. La alteración en algunos pacientes del Homeostatic Model Assessment of Insulin Resistance (HOMA-IR) se ha relacionado con un desarrollo temprano de resistencia a la insulina9. Además, se han descrito niveles bajos de masa magra, y de densidad ósea21.
Otros hallazgos clínicos menos frecuentes incluyen fisura palatina, anomalías dentales, cutis marmorata y llanto de tono grave3 (ronco).
Historia naturalLos pacientes con SCdL clásico suelen ser diagnosticados al nacer y, por lo tanto, se pueden iniciar precozmente las medidas terapéuticas adecuadas, principalmente en el área de la alimentación, siendo muchas veces necesario utilizar una sonda nasogástrica o incluso realizar una gastrostomía que puede ser necesario mantener durante los primeros meses o años de vida. Es importante tener en cuenta que los pacientes con SCdL clásico suelen nacer y crecer en percentiles por debajo de los límites normales, y por lo tanto el pediatra no debe insistir en sobrealimentarlos, lo que empeoraría el RGE que padecen la mayoría. Por ello, es recomendable utilizar las gráficas de crecimiento propias del síndrome (disponibles en: https://www.corneliadelange.es/). Las complicaciones más frecuentes a lo largo de la vida son la neumonía/neumonitis por aspiración de jugo gástrico (debido el RGE), que es una de las causas principales de mortalidad precoz14 (31% de fallecimientos), la obstrucción intestinal (debida a vólvulo en casos de malrotación intestinal) (19%), las derivadas de malformaciones congénitas graves (hernia diafragmática o cardiopatía congénita) (15%). Otras causas de mortalidad precoz fueron las complicaciones neurológicas y accidentes (8%) y la sepsis (4%).
Síndrome Cornelia de Lange no clásicoAparte del SCdL clásico, gracias a los avances en el campo del diagnóstico genético, en las últimas dos décadas se han identificado otros fenotipos «no clásicos» de SCdL, generalmente más leves, que suelen ser difíciles de diagnosticar por el pediatra. Debido a la gran variabilidad clínica de los pacientes con estas formas, algunos autores prefieren utilizar el término «espectro Cornelia de Lange» (ECdL) en lugar de SCdL.
En general, los pacientes con formas «no clásicas» suelen tener un fenotipo menos característico (rasgos no tan marcados) que los pacientes clásicos, siendo a veces lo más llamativo la sinofridia y la gestalt facial (fig. 1).
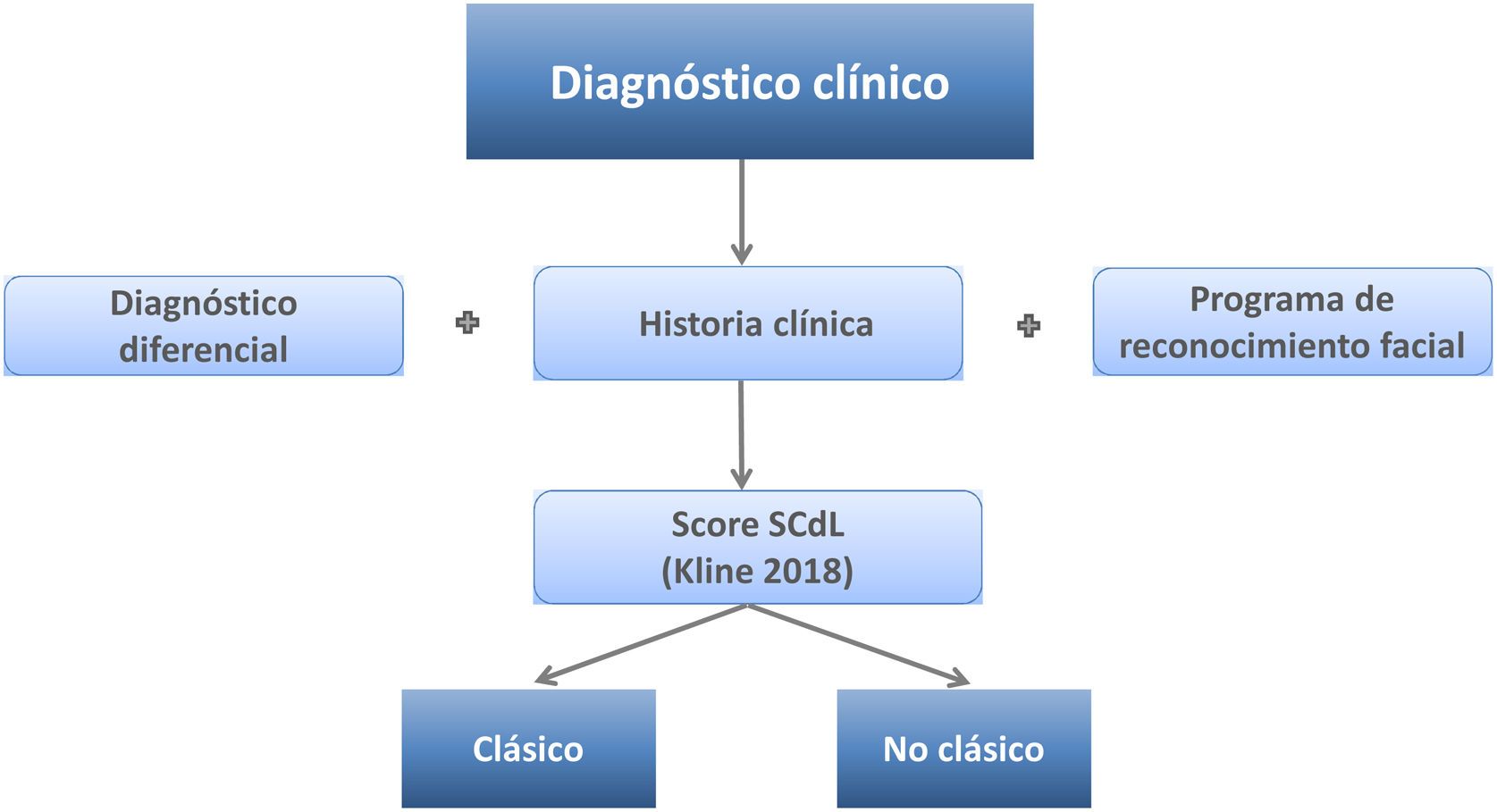
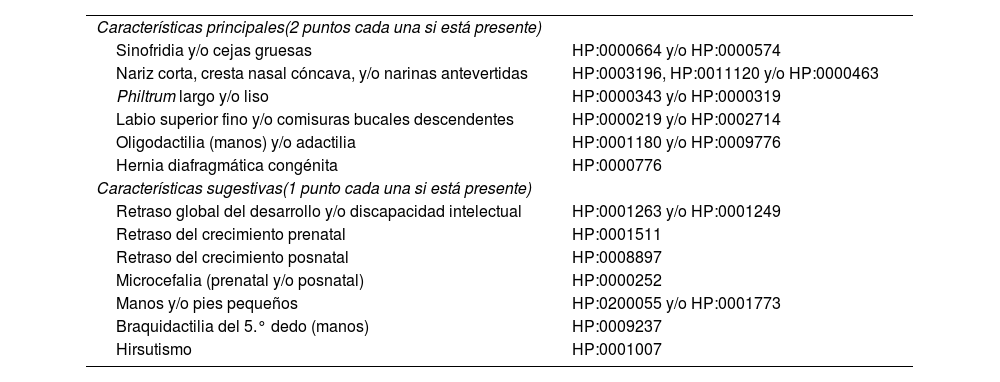
Diagnóstico diferencialEl diagnóstico diferencial del SCdL puede ser complejo debido a la variabilidad de las manifestaciones clínicas y al solapamiento con otros trastornos genéticos del neurodesarrollo, como los síndromes de Coffin-Siris, Rubinstein-Taybi, KBG, CHOPS o Wiedemann-Steiner22-26. En 2018, un consenso de expertos internacionales propuso unos criterios para el diagnóstico clínico que los pediatras pueden seguir fácilmente cuando se enfrenten a un paciente con sospecha de padecer el síndrome3 (tabla 2). Estos criterios permiten distinguir entre el fenotipo clásico y no clásico y discriminar de otras entidades similiares. Se basa en asignar un valor específico a una serie de características clínicas cardinales y a otras «sugestivas», y según la puntuación final alcanzada, clasificar en SCdL clásico (puntuación ≥11 puntos, de los cuales al menos 3 son características cardinales) y SCdL no clásico (puntuación entre 9 o 10 puntos, de los cuales al menos 2 son características cardinales). Además, en el caso de que la puntuación alcanzada esté entre 4 y 8 puntos, se justifica la realización de pruebas moleculares para determinar el diagnóstico (tabla 2).
Manifestaciones clínicas del síndrome Cornelia de Lange
| Características principales(2 puntos cada una si está presente) | |
| Sinofridia y/o cejas gruesas | HP:0000664 y/o HP:0000574 |
| Nariz corta, cresta nasal cóncava, y/o narinas antevertidas | HP:0003196, HP:0011120 y/o HP:0000463 |
| Philtrum largo y/o liso | HP:0000343 y/o HP:0000319 |
| Labio superior fino y/o comisuras bucales descendentes | HP:0000219 y/o HP:0002714 |
| Oligodactilia (manos) y/o adactilia | HP:0001180 y/o HP:0009776 |
| Hernia diafragmática congénita | HP:0000776 |
| Características sugestivas(1 punto cada una si está presente) | |
| Retraso global del desarrollo y/o discapacidad intelectual | HP:0001263 y/o HP:0001249 |
| Retraso del crecimiento prenatal | HP:0001511 |
| Retraso del crecimiento posnatal | HP:0008897 |
| Microcefalia (prenatal y/o posnatal) | HP:0000252 |
| Manos y/o pies pequeños | HP:0200055 y/o HP:0001773 |
| Braquidactilia del 5.° dedo (manos) | HP:0009237 |
| Hirsutismo | HP:0001007 |
Interpretación de la puntuación obtenida:
≥ 11 puntos, de los cuales al menos 3 son principales: SCdL clásico.
9 o 10 puntos, de los cuales al menos 2 son principales: SCdL no clásico.
4-8 puntos, de los cuales al menos 1 es principal: se indican estudios moleculares para SCdL.
<4 puntos: insuficiente para indicar estudios moleculares para SCdL.
Además, en los últimos años han surgido herramientas de inteligencia artificial (IA) que, analizando los rasgos faciales, facilitan el diagnóstico clínico de enfermedades genéticas raras. A partir de una fotografía frontal de la cara de individuos con SCdL, Latorre et al.27 testaron la aplicación de Face2Gene (https://www.face2gene.com). Los resultados mostraron que esta tecnología tenía una alta sensibilidad en el reconocimiento facial del SCdL, llegando a alcanzar el 83,7% de éxito, si se tiene en cuenta el primer síndrome sugerido (fig. 1).
En resumen, a la hora de realizar el diagnóstico (fig. 2) es importante una historia clínica detallada y estandarizada que recoja la información según los códigos Human Phenotype Ontology (HPO) (tabla 1). Además, puede ser cada vez más útil la utilización de herramientas de IA para el reconocimiento facial (fig. 1). Sin embargo, el diagnóstico clínico final, y la clasificación entre SCdL clásico o no clásico, vendrán dados por la aplicación de los criterios de Kline et al.3 (tabla 2).
Diagnóstico molecular
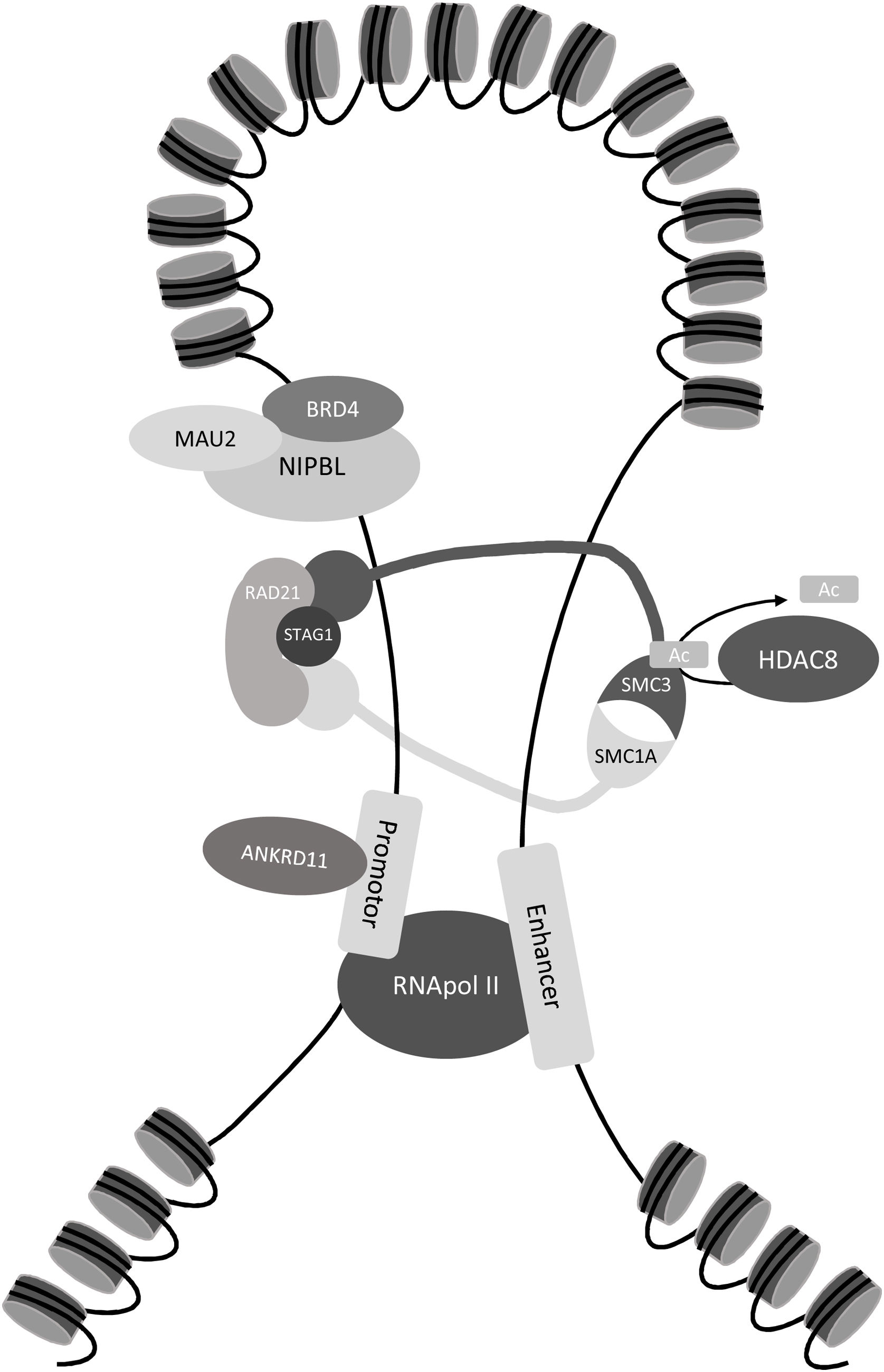
El SCdL puede diagnosticarse mediante la identificación de una variante patogénica en uno de los cinco genes causales asociados al complejo de cohesinas (NIPBL, SMC1A, SMC3, RAD21, HDAC8) y en menor medida, en tres genes adicionales descritos recientemente (BRD4, ANKRD11, MAU2)28,29. El complejo de cohesinas consta de un anillo central formado por las proteínas SMC1A, SMC3 y RAD21, y una serie de proteínas reguladoras como NIPBL, principal mediador de la carga de cohesinas en el ADN, y HDAC8, que es una deacetilasa de SMC3 que permite la disociación de cohesinas y cromatina3 (fig. 3).

Localización de las proteínas causales del síndrome Cornelia de Lange: NIPBL y BRD4 son las encargadas de eliminar los nucleosomas del ADN para que las proteínas SMC1A, SMC3, RAD21 y STAG2 puedan formar el anillo de cohesinas. MAU2 forma un heterodímero con NIPBL para ayudar a la carga de las cohesinas en la cromatina. Después la proteína HDAC8 se encargará de deacetilar a SMC3 y permitir la disociación del anillo. La proteína ANKRD11 participa en el proceso de activación de la transcripción.
El complejo de cohesinas es un complejo multifuncional que interviene en numerosos procesos biológicos, como la segregación cromosómica, el mantenimiento de la estabilidad y la organización genómica y, sobre todo, la regulación de la expresión génica. La mayoría de los individuos afectados, más del 60%, tienen una variante patogénica en NIPBL que codifica una proteína que interacciona con remodeladores de la cromatina y factores de la transcripción30. Es por ello que este síndrome se clasifica como una transcriptomopatía, es decir, una enfermedad causada por la desregulación global de la expresión génica en todo el organismo, lo que explica la afectación multisistémica y la variabilidad fenotípica del síndrome28.
Los avances tecnológicos de los últimos años en secuenciación masiva han permitido identificar nuevos genes causales, especialmente en los pacientes que presentan un fenotipo no clásico. Entre estos genes destacan ANKRD11, que codifica proteínas reguladoras de la transcripción24, o MAU2 y BDR4, que codifican proteínas que interaccionan con NIPBL29,31 (fig. 3).
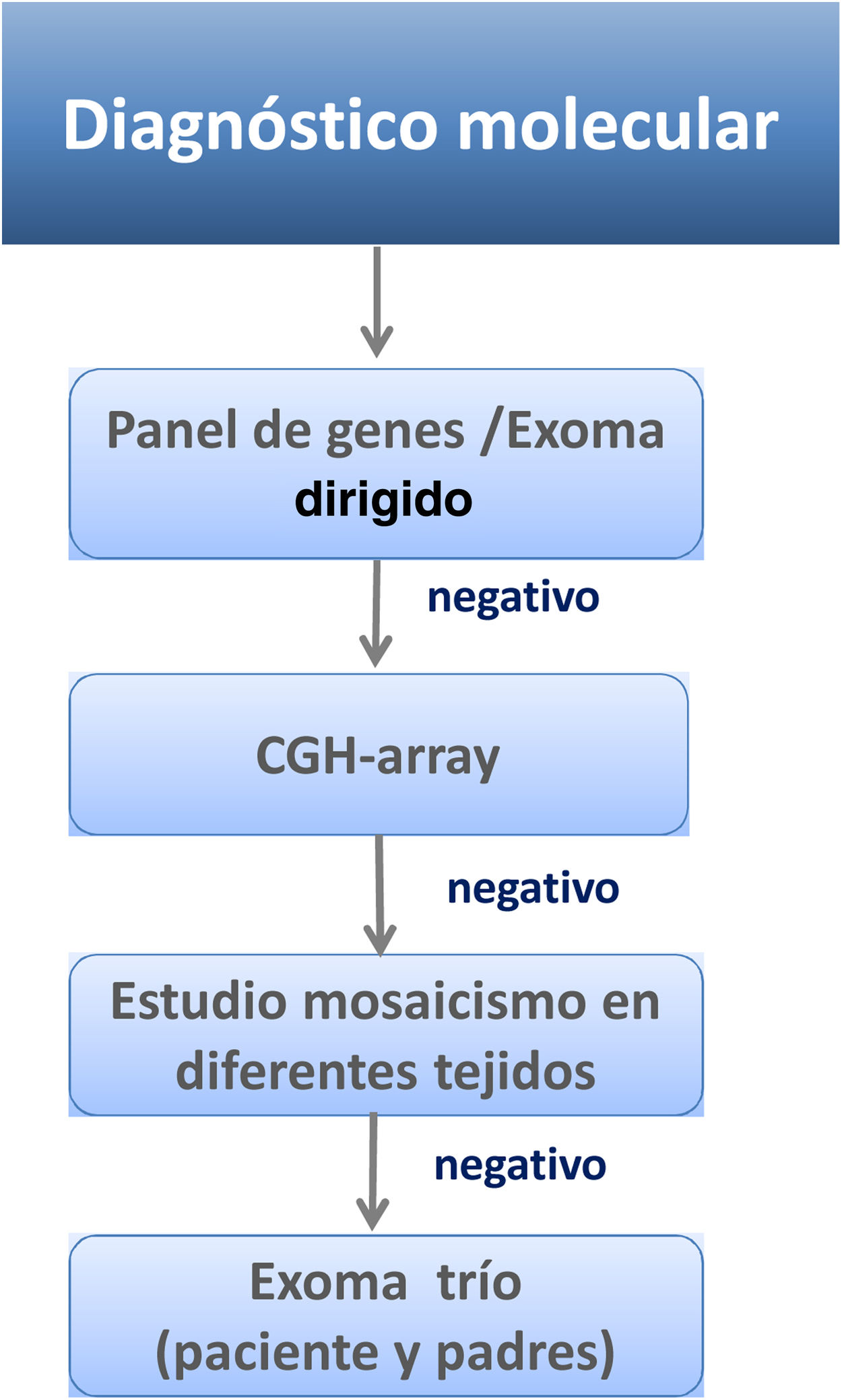
Este alto grado de heterogeneidad clínica y genética, especialmente entre los individuos con SCdL no clásico/leve o atípico, a menudo puede obstaculizar el diagnóstico molecular. Por ello, se recomienda como primera aproximación la utilización de técnicas de secuenciación masiva que permitan evaluar simultáneamente un número considerable de genes (fig. 4). La tendencia en los últimos años consiste en utilizar paneles que incluyen genes causales de la enfermedad y genes asociados a trastornos del neurodesarrollo, o exomas clínicos que evalúan simultáneamente hasta 7.000 genes asociados con patología. Bien es cierto que la mejora de las tecnologías, la reducción en los costes de la secuenciación y la automatización del análisis de datos hacen que previsiblemente en los próximos años los exomas completos (e incluso genomas) puedan sustituir estas técnicas.

Si tras la realización de todas estas pruebas obtenemos un resultado negativo, se recomienda la realización de un CGH-Array (hibridación genómica comparada) que aporte información sobre anomalías cromosómicas o variantes en el número de copias en el ADN.
La presencia de mosaicismo somático en algunos individuos (10-13%) puede representar una complicación adicional, siendo necesario el análisis en laboratorios especializados de otros tejidos, como mucosa oral o fibroblastos, y la utilización de técnicas de diagnóstico molecular con alta sensibilidad, cuando los resultados en sangre sean negativos32,33.
Si aun así no se pudiera llegar al diagnóstico, se recomienda realizar un estudio genético de exoma trío en paciente y progenitores. Las muestras de los padres han de servir para filtrar las variantes encontradas en el caso índice (fig. 4).
Sin embargo, a pesar de que se han realizado grandes progresos, sigue habiendo un número significativo de individuos sin diagnóstico genético que sugiere la existencia de genes causales y mecanismos mutacionales no identificados.
Relaciones genotipo-fenotipoAunque la mayoría de individuos con SCdL clásico tienen una variante patogénica en el gen NIPBL, que se acompaña de facies característica y, a veces, reducción de las extremidades, hay también pacientes con fenotipo no clásico (fig. 5). Las variantes patogénicas de NIPBL se distribuyen uniformemente a lo largo de la secuencia codificante, pero las variantes patogénicas de cambio de sentido (missense) son más frecuentes dentro del dominio de repetición HEAT y más leves desde el punto de vista clínico32,34.

Los individuos con variantes patogénicas en SMC1A o SMC3 tienen menos anomalías estructurales y una restricción del crecimiento menos severa que aquellos con variantes patogénicas en NIPBL; sin embargo, tienen una discapacidad intelectual significativa que puede variar de moderada a grave35. Los pacientes con variantes patogénicas en SMC1A se acompañan de cejas ligeramente más planas y anchas, y puente nasal amplio y largo; en algunas ocasiones presentan epilepsia intratable de inicio temprano36. Aquellos con variantes patogénicas en SMC3 a menudo tienen sinofrias sutiles o ausentes; además, en el 56% de los casos se observan malformaciones cardíacas37.
Los individuos con variante patogénica heterocigótica en el gen RAD21 generalmente no tienen alteraciones estructurales importantes; además, el deterioro cognitivo y el RGE tienden a ser más leve, limitándose este último a la primera infancia38. Estos individuos suelen mostrar retraso en el crecimiento, anomalías esqueléticas menores y rasgos faciales que se superponen con el SCdL.
Los varones con variante patogénica hemicigótica en HDAC8 tienen rasgos faciales típicos del síndrome, pero acompañados de un cierre tardío de la fontanela anterior, párpados caídos, ojos muy espaciados, pigmentación de la piel en mosaico, anomalías dentales y, sobre todo, una punta nasal ancha y bulbosa con una personalidad amistosa. El crecimiento está menos afectado, siendo menos frecuente el retraso posnatal y la microcefalia. En algunas ocasiones pueden presentar también reducción grave de las extremidades. En las mujeres, el cuadro tiende a ser más leve, pero la gravedad de la presentación clínica está muy influenciada por el patrón de inactivación del cromosoma X39.
Asesoramiento genéticoLa mayoría de los casos de SCdL son esporádicos, especialmente los asociados a los genes cuya herencia sigue un patrón autosómico dominante (AD): NIPBL, SMC3 y RAD21. En pacientes con variantes patogénicas en genes ligados alX (LX) (SMC1A y HDAC8) los varones suelen estar más afectados que las mujeres. En estas familias hay casos de progenitores levemente afectados y el riesgo de transmisión dependería del sexo del progenitor3.
Más del 99% de los pacientes con SCdL clásico tienen una variante patogénica heterocigótica no heredada (de novo) en el gen NIPBL. Esto supone que el riesgo de recurrencia en futuros hermanos es muy bajo (1-2%), debido principalmente a la posibilidad de mosaicismo en la línea germinal de uno de los progenitores40. En las familias en las que se ha identificado la variante patogénica en cualquiera de los genes relacionados con el SCdL sería posible realizar el estudio genético prenatal para identificar o descartar dicha variante en el feto.
Tratamiento y seguimientoHoy día no hay tratamiento curativo para el SCdL, pero sí tratamientos sintomáticos/paliativos que deben ser conocidos por el pediatra. La principal complicación médica en el SCdL clásico (asociado al gen NIPBL) es el RGE, presente en la gran mayoría de los pacientes, que debe ser tratado de forma precoz y contundente, siguiendo los protocolos terapéuticos validados en la literatura pediátrica. En ocasiones, ante una respuesta ineficaz al tratamiento médico estándar, es preciso el empleo de medidas alternativas, como la alimentación por sonda nasogástrica o por gastrostomía durante los primeros meses/años de vida. Por último, en algunos pacientes es necesario recurrir al tratamiento quirúrgico (funduplicatura de Nissen). En relación con el aparato digestivo es importante tener en cuenta la posibilidad de que exista una malrotación intestinal que puede complicarse con un cuadro de dolor abdominal agudo (por obstrucción o vólvulo), constante o recurrente, que lleva al paciente (generalmente en los dos primeros años de vida) al servicio de urgencias del hospital, donde si no se identifica la causa precozmente, puede producirse un desenlace fatal3.
Es obligado comprobar la audición desde el nacimiento (otoemisiones acústicas); si hubiera dudas o fueran negativas, es necesaria la realización de potenciales evocados auditivos corticales (PEAC) para, en caso de hipoacusia neurosensorial, planear la colocación de dispositivos de ayuda auditiva (audífono o implante coclear) lo antes posible, preferiblemente antes del inicio del lenguaje oral. Asimismo, debe realizarse un ecocardiograma y una ecografía abdominal para comprobar si existen anomalías estructurales en el corazón o en alguno de los órganos abdominales (riñones).
Desde el punto de vista del desarrollo psicomotor, en la mayoría de los pacientes con SCdL será necesario el empleo de técnicas de estimulación del lenguaje (logopedia) y del desarrollo motor (terapia física y/o terapia ocupacional). En algunos casos puede ser necesario el empleo de fármacos para la hiperactividad y/o déficit de atención.
El pasado mes de noviembre de 2023 se celebró en Zaragoza en XIICongreso Mundial del SCdL, organizado por el Centro de Referencia del SCdL en colaboración con la Asociación Española y la Federación Mundial del SCdL. A dicho Congreso acudieron los principales expertos mundiales, que compartieron los últimos avances en el conocimiento del síndrome. En las áreas clínicas cabe destacar la novedosa técnica de la «neuroestimulación sensible» para tratar la epilepsia refractaria de los pacientes con SCdL con variantes patogénicas en el gen SMC1A, cuya eficacia se ha demostrado en 3 de cada 4 pacientes tratados. También se habló de la importancia de considerar la presencia de una malrotación intestinal en todo niño con SCdL que acude a urgencias por dolor abdominal agudo. En relación con los procedimientos anestésicos en niños con SCdL, se insistió en que la experiencia ha ratificado su buena tolerancia a los mismos. En las áreas de investigación básica se confirmó que la ausencia de cohesina da lugar a alteraciones en la transcripción del ADN; se expuso la obtención de novedosos modelos 3D de cohesina, potencialmente útiles para el descubrimiento de potenciales dianas terapéuticas en el SCdL. También se está trabajando en modelos animales para iniciar ensayos clínicos con Ataluren, para el tratamiento de la epilepsia en pacientes con SCdL-SMC1A. Se presentaron estudios en célula única que demostraron la existencia de disregulación transcripcional en las primeras etapas del desarrollo embrionario en modelo de ratón. Por último, se presentaron los primeros resultados de estudios de senescencia celular en SCdL utilizando células madre pluripotenciales derivadas de pacientes con SCdL.
FinanciaciónPrograma AES 2023 (ISCiii, Ministerio de Ciencia, Innovación y Universidades): Proyecto PI23/01370; y Diputación General Aragón: Grupo Investigación B32-20R.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

