

La enfermedad por inclusiones microvellositarias es una entidad rara, de herencia autosómica recesiva y caracterizada por una diarrea grave de carácter secretor que produce un fracaso intestinal permanente dependiente de nutrición parenteral. Habitualmente se inicia en el período neonatal y el único tratamiento posible en el momento actual es el trasplante intestinal.
Pacientes y métodosSe revisa, de forma retrospectiva, a 6 pacientes (3 varones y 3 mujeres), diagnosticados entre 1998 y 2013 de enfermedad por inclusiones microvellositarias.
ResultadosTodos comenzaron en el primer mes de vida, con una mediana de edad de tres días (rango: 3-30 días) y presentaron diarrea secretora dependiente de nutrición parenteral, con un volumen fecal en ayunas de 150-200ml/kg/día. La microscopia óptica de muestras biópsicas duodenales mostró grados variables de atrofia vellositaria sin hiperplasia críptica, con acumulación de material PAS positivo en el citoplasma de los enterocitos del borde en cepillo y la inmunotinción anti-CD10 fue indicativa de inclusiones intracitoplasmáticas. La confirmación diagnóstica se realizó con microscopia electrónica. En 2 de ellos se realizó estudio genético que demostró mutaciones en el gen MYO5B. Evolutivamente, 3 fallecieron y 3 se encuentran vivos; 2 de ellos portadores de trasplante intestinal y el tercero en espera de trasplante multivisceral.
Microvillous inclusion disease is a rare autosomal recessive condition, characterized by severe secretory diarrhea that produces a permanent intestinal failure and dependency on parenteral nutrition. It usually begins in the neonatal period, and the only treatment at present is intestinal transplantation.
Patients and methodsA retrospective review was conducted on 6 patients (three males and three females) diagnosed with microvillous inclusion disease between 1998 and 2013.
ResultsAll debuted in the first month of life, with a median age of three days (range, 3-30 days), and had secretory diarrhea dependent on parenteral nutrition, with fasting fecal volume of 150-200ml/kg/day. Light microscopy of duodenal biopsy samples showed varying degrees of villous atrophy without cryptic hyperplasia, accumulation of PAS positive material in the cytoplasm of enterocytes brush border, and anti-CD10 immunostaining was suggestive of intracytoplasmic inclusions. Diagnostic confirmation was performed with electron microscopy. Two of them had a genetic study, and showed mutations in MYO5B gene. Three died and three are alive; two of them with an intestinal transplantation and the third waiting for a multivisceral transplantation.
La enfermedad por inclusiones microvellositarias (EIM) o atrofia microvellositaria congénita/familiar es debida a un trastorno congénito del enterocito. Se han descrito múltiples mutaciones en el gen MYO5B, que codifica la miosina Vb, implicadas en el desarrollo de esta enfermedad. Se presenta típicamente en el período neonatal con una diarrea grave que persiste a pesar del ayuno, dependiente de nutrición parenteral (NP) de forma definitiva. Histológicamente, se caracteriza por la ausencia o alteración del ribete en cepillo del enterocito junto a la presencia de inclusiones microvellositarias características. El tratamiento incluye la NP y el trasplante intestinal.
Pacientes y métodosSe revisó de forma retrospectiva a los pacientes diagnosticados de EIM en nuestro hospital en un período de 15 años (1998-2013). Se analizaron variables epidemiológicas, clínicas, diagnósticas, terapéuticas y evolutivas. Se realizó endoscopia digestiva alta con toma de biopsias de duodeno para la confirmación diagnóstica. Se practicó un examen histológico convencional con tinciones de hematoxilina-eosina (HE) y ácido peryódico de Schiff (PAS), así como inmunohistoquímica con anticuerpos anti-CD10 y estudio con microscopía electrónica. Se realizó análisis genético en 2 de los 6 pacientes, para estudio de mutaciones del gen MYO5B. En todos ellos se descartaron causas infecciosas, alérgicas, autoinmunes, trastornos de digestión, absorción y transporte de nutrientes, y otras enfermedades de la mucosa intestinal.
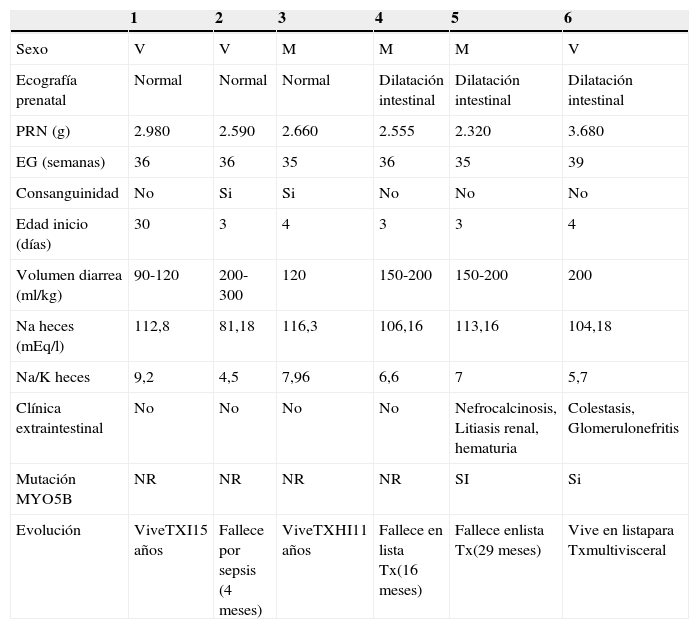
ResultadosLa diarrea se inició durante los 4 primeros días de vida en 5 pacientes, con una mediana de edad de 3 días (rango 3-30 días) (tabla 1). Tres eran varones y 3 mujeres. Los pacientes 2 y 3 eran hermanos y nacidos de padres consanguíneos, no existiendo ningún tipo de parentesco entre los progenitores del resto de los pacientes. El paciente 6 fue resultado de una gestación que llegó a término, mientras el resto tuvo una edad gestacional comprendida entre 35 y 36 semanas. Todos presentaron al nacimiento un peso adecuado para la edad gestacional. Las ecografías prenatales mostraron, en la mitad de los pacientes, la existencia de dilatación de asas intestinales sin otros hallazgos significativos.
Características generales de los pacientes con enfermedad por inclusiones microvellositarias
| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| Sexo | V | V | M | M | M | V |
| Ecografía prenatal | Normal | Normal | Normal | Dilatación intestinal | Dilatación intestinal | Dilatación intestinal |
| PRN (g) | 2.980 | 2.590 | 2.660 | 2.555 | 2.320 | 3.680 |
| EG (semanas) | 36 | 36 | 35 | 36 | 35 | 39 |
| Consanguinidad | No | Si | Si | No | No | No |
| Edad inicio (días) | 30 | 3 | 4 | 3 | 3 | 4 |
| Volumen diarrea (ml/kg) | 90-120 | 200-300 | 120 | 150-200 | 150-200 | 200 |
| Na heces (mEq/l) | 112,8 | 81,18 | 116,3 | 106,16 | 113,16 | 104,18 |
| Na/K heces | 9,2 | 4,5 | 7,96 | 6,6 | 7 | 5,7 |
| Clínica extraintestinal | No | No | No | No | Nefrocalcinosis, Litiasis renal, hematuria | Colestasis, Glomerulonefritis |
| Mutación MYO5B | NR | NR | NR | NR | SI | Si |
| Evolución | ViveTXI15 años | Fallece por sepsis (4 meses) | ViveTXHI11 años | Fallece en lista Tx(16 meses) | Fallece enlista Tx(29 meses) | Vive en listapara Txmultivisceral |
M: mujer; NR: no realizado; TXHI: trasplante hepatointestinal; TXI: trasplante intestinal; V: varón.
Los 6 comenzaron con una diarrea grave (volumen fecal en torno a 120-200ml/kg/día) de carácter secretor (sodio en heces mayor de 100 mEq/l), con abundante moco y persistencia de la diarrea pese al ayuno. La paciente 5 desarrolló complicaciones renales en relación con la NP y los graves disturbios hidroelectrolíticos y el paciente 6 presentó prurito cutáneo intenso como síntoma muy destacado. No fue tolerado ningún tipo de fórmula por vía enteral, precisando todos ellos NP para evitar la deshidratación y la acidosis metabólica.
La endoscopia digestiva alta no mostró alteraciones macroscópicas significativas. El diagnóstico se realizó mediante el estudio histológico de muestras de biopsias duodenales con microscopía óptica y electrónica. En los pacientes 5 y 6 se confirmó la existencia de mutaciones en el gen MYO5B. No se realizó estudio genético en el resto de los pacientes. Todos ellos recibieron NP hospitalaria/domiciliaria durante períodos prolongados (3-36 meses). Tres de los 6 pacientes han fallecido (uno a los 3 meses de vida por shock séptico y los otros 2 en lista de espera de trasplante por complicaciones hepáticas asociadas al fracaso intestinal). De los 3 supervivientes, uno se encuentra en espera de un trasplante multivisceral y ha desarrollado una glomerulonefritis aguda postinfecciosa y los otros 2 están trasplantados (uno intestino aislado y el otro hepatointestinal), hace 15 y 11 años, respectivamente, con autonomía digestiva y buena calidad de vida.
DiscusiónExisten distintas causas de diarrea congénita de inicio en los primeros meses de vida: defectos en la digestión, absorción y transporte de nutrientes y electrolitos, defectos en la diferenciación y polarización de los enterocitos, defectos en la diferenciación de la célula enteroendocrina y defectos en la modulación de la respuesta inmunitaria intestinal1. Dentro de los trastornos congénitos del enterocito, destacan fundamentalmente la displasia epitelial intestinal y la EIM, que son causas infrecuentes de fracaso intestinal permanente.
La EIM, también denominada atrofia microvellositaria congénita, fue descrita por primera vez en 1978 en 5 pacientes por Davidson et al.2, siendo el nombre de EIM propuesto por Cutz et al. en 19893. Se trata de una enfermedad congénita en la que recientemente se han descrito mutaciones en el gen MYO5B localizado en el cromosoma 18, que codifica una expresión alterada de la miosina Vb en los enterocitos4-7. La miosina Vb es una proteína que, mediante la unión con proteínas Rab, proporciona una base molecular para la regulación de las vías de reciclaje de los endosomas y su transporte a través de los filamentos de actina hasta la membrana apical en las células epiteliales polarizadas. Se han caracterizado mutaciones en homocigosis y otras en heterocigosis capaces de producir EIM4-7. La alteración genética exacta se desconocía hasta el año 20084,5. Posteriomente, se han descrito distintas mutaciones en el gen MYO5B, identificándose hasta 41 mutaciones distintas en el momento actual8. En los 2 pacientes más recientes de nuestra serie de realizó estudio genético con análisis del gen MYO5B, identificándose en ambos 2 mutaciones en heterocigosis; en el paciente 5 las mutaciones c.3190C>T (p.R1064X) y c.3514C>T (p.Q1172X) y en el paciente 6 las mutaciones c.445C>T (p.Q149X) y c.5383C>T (p.R1795X). No se realizó el estudio genético en los pacientes diagnosticados previamente.
Existen pacientes con EIM en los que no se ha podido demostrar mutación en el gen MYO5B. Muy recientemente, la secuenciación del exoma de ADN de pacientes con EIM ha mostrado en algunos de ellos la existencia de mutaciones en homocigosis de la sintaxina 39. La sintaxina 3 es un receptor implicado en la fusión de membranas de vesículas apicales en los enterocitos, produciéndose un acoplamiento defectuoso de las mismas a la membrana apical. Se ha postulado como una variante atípica de EIM, con expresión fenotípica más leve, con una aparición más tardía, mayor tolerancia a la alimentación enteral y sutiles diferencias histológicas10. Sin embargo, hacen falta más estudios para poder correlacionar adecuadamente cómo afectan las distintas mutaciones a la expresión fenotípica.
Durante el embarazo, aunque de forma inconstante, puede observarse por ecografía la presencia de polihidramnios y dilatación de asas intestinales11. Tres de nuestros pacientes presentaban en la ecografía del tercer trimestre dilatación intestinal, sin polihidramnios. En el resto de los casos, las ecografías prenatales fueron normales.
La enfermedad se manifiesta clínicamente por una diarrea intratable que típicamente se inicia en la primera semana de vida (forma de presentación precoz), aunque se han descrito también casos con un inicio tras las primeras 6-8 semanas de vida (forma de presentación tardía)12–14. Se trata de una diarrea líquida y abundante con gran cantidad de moco, que puede confundirse en ocasiones con la orina, con volúmenes entre 100-200ml/kg/día a pesar de mantener al paciente en ayuno. Produce una deshidratación importante en pocas horas, precisando altos aportes hidroelectrolíticos y NP de forma permanente. Todos nuestros pacientes presentaban al inicio pérdida significativa del peso con acidosis metabólica y abundante pérdida de sodio y cloro en las heces (en torno a 100mmol/l y 80mmol/l, respectivamente). La paciente 5 se remitió desde otro hospital orientada inicialmente como una posible fístula entero-vesical, dadas las características de las deposiciones.
Clásicamente, se ha considerado que la EIM no suele asociarse a otras malformaciones extraintestinales, pero algunos autores describen la coexistencia en algunos de sus pacientes de malformaciones cardíacas, síndrome de Down, displasia neuronal, hipocondroplasia o deficiencias sensoriales entre otras14–19. A pesar de la amplia expresión del gen MYO5B en todos los tejidos epiteliales, no está claro en qué forma otros órganos distintos del intestino pueden verse afectados en esta enfermedad. Durante los últimos años, se ha comprobado que las manifestaciones extraintestinales no son tan infrecuentes como se pensaba. En una serie de 24 pacientes con EIM en seguimiento desde el nacimiento hasta los 23,5 años (mediana de seguimiento de 4,7 años), publicada recientemente, la presencia de enfermedad hepática se registró en 22 pacientes, la enfermedad renal en 9 y la enfermedad pulmonar en 2 pacientes19.
La enfermedad hepática, incluyendo colestasis, colelitiasis y fibrosis hepática, puede ser consecuencia de la NP prolongada. Sin embargo, los niveles ligeramente elevados o normales de gammaglutamiltranspeptidasa (GGT) indican que la NP puede no ser la única causa de la disfunción hepática. Se ha descrito una enfermedad hepática específica asociada a EIM con GGT normal, similar a la colestasis intrahepática familiar progresiva (CIFP)19. La miosina Vb es necesaria para la formación canalicular de la bilis y también para el tráfico de los transportadores que tienen un dominio de unión a ATP20. Girard et al.21, en una cohorte de 28 pacientes con EIM, en la que 8 desarrollan colestasis antes o después del trasplante, con unas características clínicas y analíticas similares a la CIFP, concluyen que la colestasis de estos pacientes podría estar en relación con la alteración de la vía de reciclaje de endosomas MYO5B/Rab11a en los hepatocitos, la expresión alterada de la bomba exportadora de sales biliares (BSEP) en la membrana canalicular y a un aumento de la absorción ileal de ácidos biliares. La razón por la cual no todos desarrollan colestasis se desconoce. Nuestro paciente 6 presentó durante 9 meses una colestasis significativa con prurito cutáneo intenso y GGT normal. Se realizó biopsia hepática que no mostró alteraciones histológicas significativas, y se descartó por inmunohistoquímica la existencia de un déficit de BSEP y de MDR3. Actualmente se ha solucionado el cuadro colestático y se encuentra como candidato a trasplante multivisceral. En los pacientes con afectación hepática pretrasplante, se ha propuesto el trasplante hepatointestinal para evitar el desarrollo de síndromes colestáticos postrasplante.
Se ha descrito también asociación con síntomas renales, como la hematuria o el síndrome de Fanconi22, y síntomas endocrinológicos, como la diabetes23. La paciente 5 desarrolló nefrocalcinosis, litiasis y hematuria, y el paciente 6 una glomerulonefritis aguda postinfecciosa hipocomplementémica.
El diagnóstico se realiza mediante la demostración en biopsia duodenal o yeyunal de los hallazgos histológicos típicos de esta enfermedad. Aunque la microscopia óptica orienta, el diagnóstico definitivo debe realizarse con microscopia electrónica24,25. Estas alteraciones histológicas pueden afectar igualmente al colon13 o a la mucosa gástrica26. La presencia de mutaciones del gen MYO5B completa el diagnóstico.
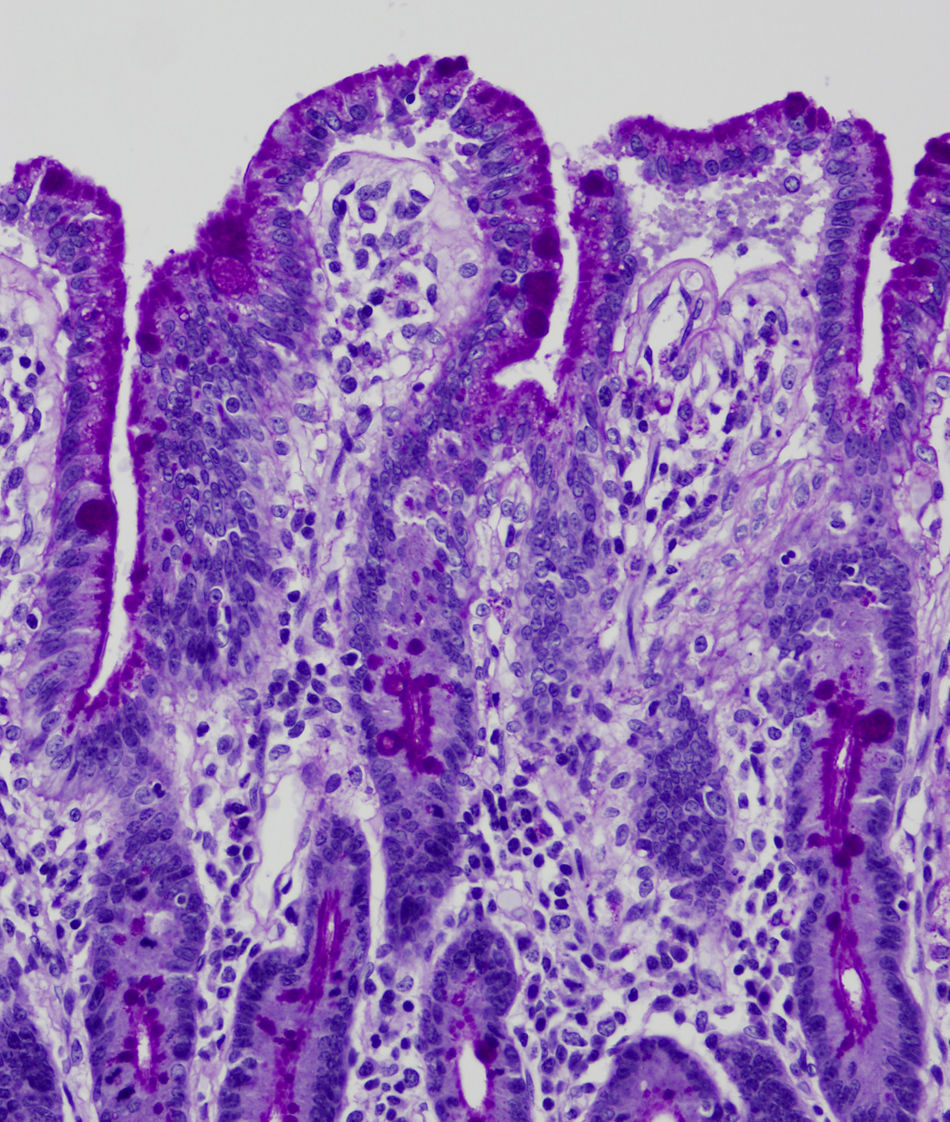
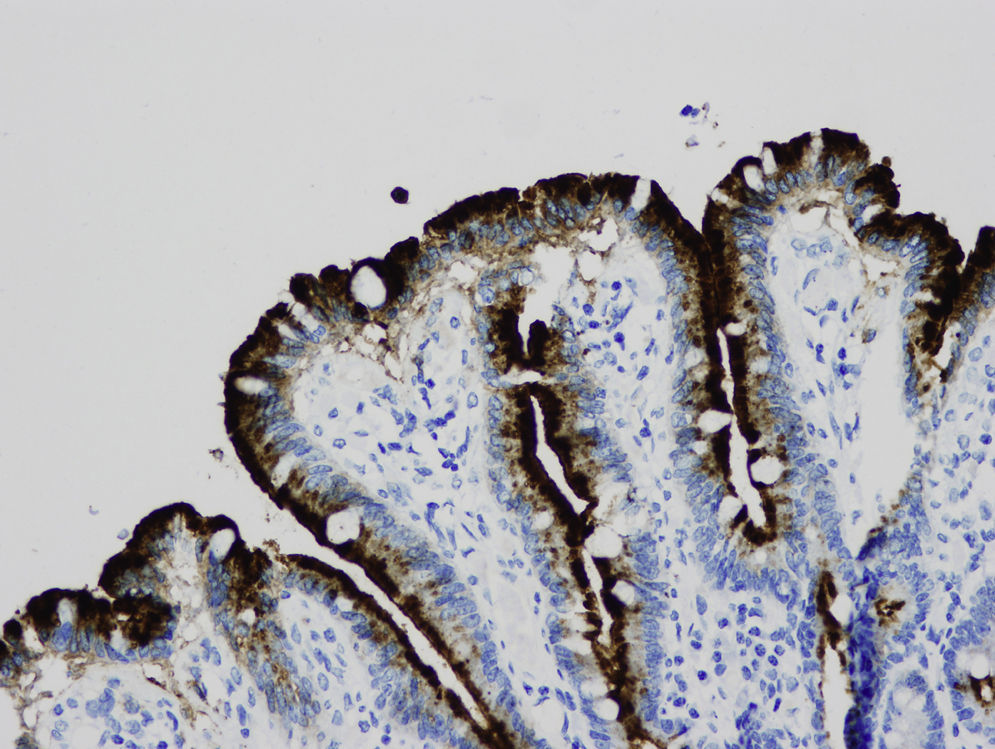
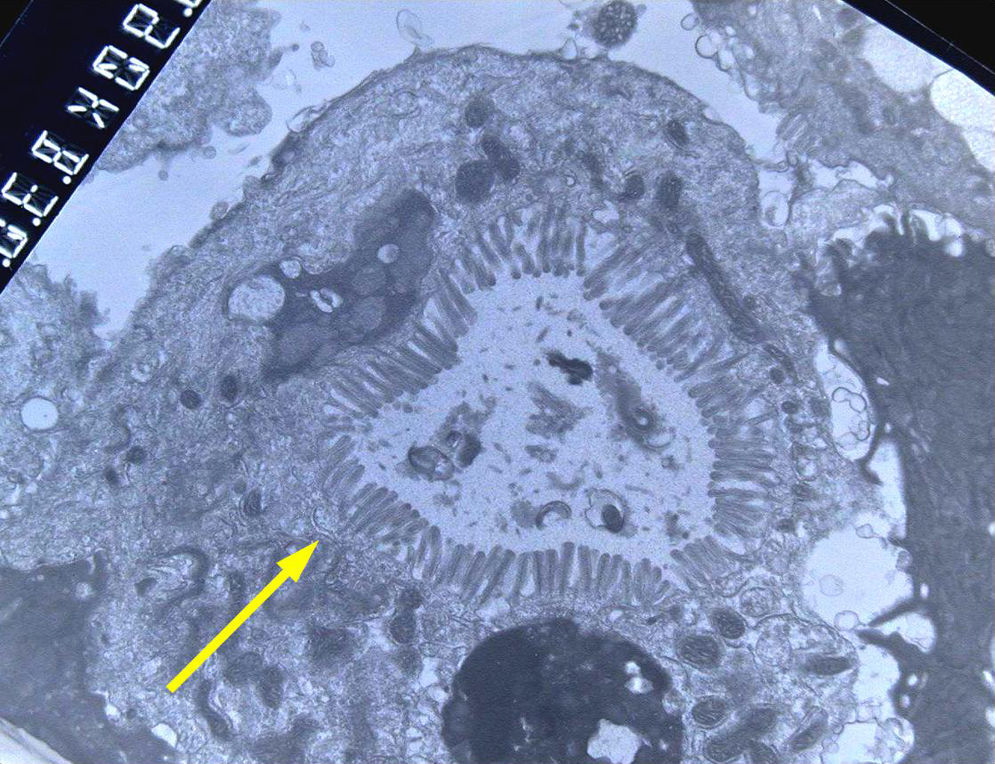
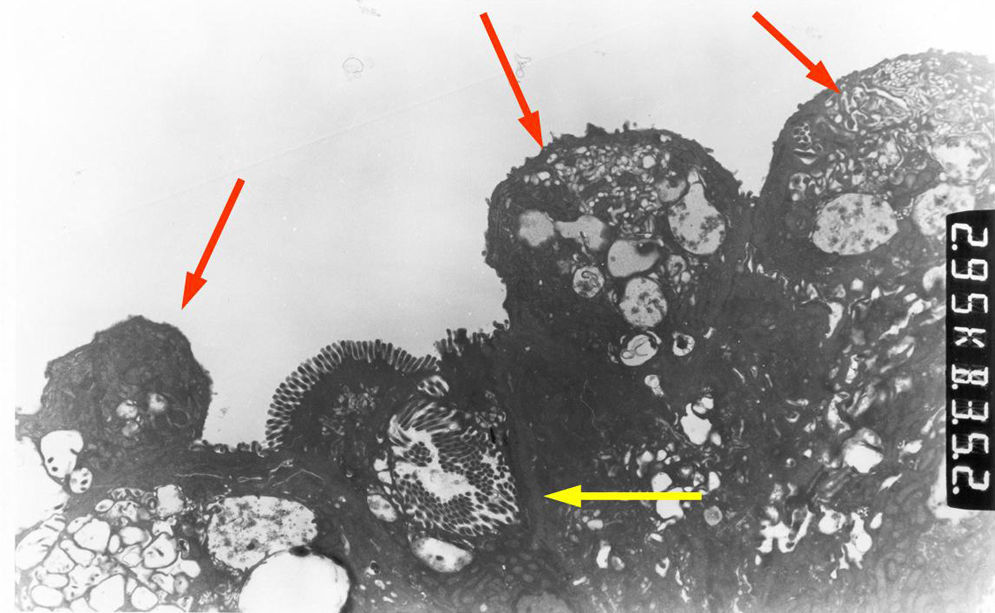
En la microscopia óptica se visualiza una atrofia variable de las vellosidades del borde en cepillo, sin reacción inflamatoria significativa y con densidad normal o levemente aumentada de las criptas. La tinción con fosfatasa alcalina27 y PAS en pacientes sanos tiñe exclusivamente el borde en cepillo del enterocito, mientras que en la EIM no tiñe o lo hace de forma irregular a este nivel, presentándose una acumulación de material con positividad PAS en el citoplasma apical de los enterocitos (fig. 1). Este hecho puede ponerse también de manifiesto con inmunohistoquímica frente a CD1028 (fig. 2) y antígeno carcinoembrionario29. Los 3 hallazgos ultraestructurales fundamentales son: la disminución, la ausencia y/o la desorganización de microvellosidades de los enterocitos maduros; la acumulación, en el citoplasma apical, preferentemente de células de la cripta, de gránulos secretores y los característicos cuerpos de inclusión intracitoplasmáticos con microvellosidades en su cara interna (fig. 3). Sin embargo, no siempre es fácil el diagnóstico anatomopatológico de la EIM por la existencia de variantes, distintos comportamientos clínicos e incluso la existencia de un pequeño porcentaje de pacientes en los que no se consiguen demostrar los cuerpos de inclusión típicos. En 1994 Raafat et al.30 describe a 3 niños con distrofia microvellositaria, que considera una variante anatomoclínica de la EIM, a la que harán referencia posteriormente otros autores24. Esta lesión ultraestructural se observó en el paciente 1 (fig. 4).




Evolutivamente, precisan de forma invariable soporte con NP, dado que la alimentación por vía enteral no es posible en relación con la gran cantidad de pérdidas digestivas con riesgo de deshidratación y fallecimiento.
Se han probado diferentes opciones terapéuticas con distintos fármacos incluyendo loperamida, somatostatina, corticoides o factores de crecimiento entre otros, pero ninguno de ellos ha sido eficaz en estos pacientes30,31. El trasplante intestinal es actualmente la única alternativa en aquellos pacientes con fracaso intestinal permanente, sobre todo en los que existen dificultades para la administración de la NP (pérdida de accesos venosos por trombosis del sistema venoso profundo) o complicaciones serias derivadas de la misma (hepatopatía, sepsis relacionadas con la presencia de un catéter central, etc.)19,32,33. Dos de nuestros pacientes han sido trasplantados con una evolución favorable, presentando en la actualidad autonomía digestiva sin haber presentado complicaciones relacionadas con su enfermedad de base. Un tercer paciente se encuentra en situación de espera de un trasplante multivisceral, aunque en la actualidad se encuentra en exclusión temporal de lista de trasplante por un cuadro de glomerulonefritis.
Se han descrito casos aislados en la literatura en los que se observó mejoría del cuadro clínico que permitió evolutivamente la autonomía digestiva con retirada de la NP16,34. Perry et al.14 en una reciente revisión de 8 casos de EIM con manifestaciones atípicas, por aparición más tardía de la clínica y con volumen fecal de menor cuantía, describen 3 casos que evolucionan favorablemente permitiendo la retirada de la NP. Por esto, proponen ampliar el espectro clínico de esta enfermedad a pacientes con variantes más leves, por lo que la EIM debe considerarse en el diagnóstico diferencial de diarrea intratable fuera del período neonatal.
Las causas de fallecimiento más habituales son descompensaciones metabólicas graves o complicaciones derivadas de la NP (hepatopatía o sepsis), como ocurrió en los 3 pacientes fallecidos de nuestra serie.
En resumen, la EIM es una enfermedad genética hereditaria grave, que produce un fracaso intestinal de inicio precoz dependiente de NP y cuyo único tratamiento curativo es el trasplante intestinal. El diagnóstico es clínico, histológico, ultraestructural y genético, pero, en ocasiones, puede resultar difícil por tratarse de cuadros atípicos o incompletos. Debe realizarse el diagnóstico diferencial con otras causas de diarrea grave de comienzo precoz.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

