


El incremento universal de la prevalencia de obesidad en niños y adolescentes durante las últimas décadas, junto con la evidencia creciente de que el establecimiento de obesidad en etapas precoces de la vida, está asociado con un incremento de la prevalencia de comorbilidades y del riesgo de muerte prematura, con gran repercusión económica en los sistemas sanitarios de los países occidentales, y ha impulsado la investigación en esta área. Estos estudios han remarcado la importante actividad endocrina del tejido adiposo, ejercida por medio de la síntesis y secreción de un gran número de péptidos y citoquinas, denominados adipoquinas.
En esta revisión se resume el estado actual de los conocimientos, así como los estudios más relevantes, en relación con la dinámica de secreción de las principales adipoquinas en niños, centrándose en el control de la homeostasia energética, la regulación metabólica (fundamentalmente el metabolismo de los hidratos de carbono) y la inflamación. Así mismo, se analizan las particularidades de la síntesis, secreción y acciones de las adipoquinas desde el nacimiento hasta la adolescencia, reseñando el efecto que, sobre ellas, ejerce la instauración de la obesidad.
The worldwide increase in the prevalence of obesity in children and adolescents during the last decades, as well as the mounting evidence indicating that obesity is associated with an increased incidence of comorbidities and the risk of premature death, resulting in a high economic impact, has stimulated obesity focused research. These studies have highlighted the prominent endocrine activity of adipose tissue, which is exerted through the synthesis and secretion of a wide variety of peptides and cytokines, called adipokines.
This review presents a summary of the current knowledge and most relevant studies of adipokine dynamics and actions in children, focusing on the control of energy homeostasis, metabolic regulation (particularly carbohydrate metabolism), and inflammation. The particularities of adipose secretion and actions in healthy children, from birth to adolescence, and the modifications induced by early onset obesity are highlighted.
Actualmente no existe una definición universalmente aceptada para el diagnóstico de obesidad en niños y adolescentes, debido al empleo de referencias poblacionales de índice de masa corporal (IMC) y puntos de corte diferentes para establecer el límite que conlleva la existencia de una acumulación patológica de tejido adiposo (TA)1. Sin embargo, sí que existe consenso respecto a dos hechos epidemiológicos: a) que la acumulación excesiva de TA en edades tempranas de la vida conlleva un incremento de la incidencia de comorbilidades presentes y futuras y del riesgo de muerte prematura2–4, y b) que la prevalencia de obesidad en niños y adolescentes se ha incrementado sustancialmente en todo el mundo en las últimas décadas5.
Estas evidencias, añadidas al impacto económico que representa la asistencia y el tratamiento de la obesidad y sus comorbilidades asociadas, para los distintos sistemas nacionales de salud, han determinado un incremento de los esfuerzos investigadores dirigidos a la obesidad y, particularmente, al mejor entendimiento de la biología del TA. Uno de los resultados de esta actividad investigadora ha sido el mejor conocimiento de la importante actividad endocrina del TA, ejercida por medio de la síntesis y secreción de una gran cantidad de péptidos y citoquinas, denominados adipoquinas6–8.
Como ocurre en muchas otras situaciones fisiopatológicas, es preciso tener en cuenta que la obesidad de instauración en edades tempranas de la vida presenta múltiples particularidades que la diferencian de la obesidad del adulto. Dichas particularidades se extienden también a la síntesis y secreción de adipoquinas tanto el periodo fetal, como en la infancia y en la adolescencia, 3 periodos de la vida caracterizados por una intensa tasa de crecimiento y desarrollo, así como por un mayor grado de plasticidad, tanto estructural como funcional de los tejidos, en comparación con la observada en la vida adulta1,9.
La lista de adipoquinas conocidas (en continuo aumento), extiende su influencia a la casi totalidad de los estados patológicos, haciendo imposible pormenorizar todos y cada uno de los péptidos secretados por el TA y detallar sus múltiples acciones. Así pues, en la presente revisión recopilaremos el estado actual del conocimiento de las acciones endocrinas del TA, centrándonos en las adipoquinas que ejercen un papel más destacado en la regulación de la homeostasia energética, de la regulación metabólica (particularmente de los hidratos de carbono) y de la inflamación. Destacando las particularidades en la secreción y acciones de estas adipoquinas en niños sanos, desde el nacimiento hasta la finalización de su desarrollo puberal y evaluando las modificaciones que, sobre las mismas, ejercen tanto el desarrollo de la obesidad, como la reducción ponderal.
El tejido adiposo blanco como órgano endocrino. Eje hipotálamo-hipofisario-adiposoEl TA consta de 2 componentes esenciales, los adipocitos y la matriz estromo-vascular. Los adipocitos constituyen la célula específica del mismo, siendo la acumulación de lípidos su capacidad fundamental. La fracción estromo-vascular está compuesta por una matriz de colágeno, nervios, sangre y vasos linfáticos, en la que se encuentran múltiples subpoblaciones celulares, como fibroblastos, preadipocitos y células del sistema mononuclear-fagocítico (SMF), principalmente monocitos y macrófagos.
A las 6 semanas de gestación, ya es posible la identificación de adipocitos en el embrión humano10,11. En este momento, la mayor parte de la grasa se acumula en forma de TA marrón (TAM), con unos adipocitos «diseñados» fundamentalmente para el consumo de energía y la generación de calor (termogénesis), en contraposición a los del TA blanco (TAB), destinados al almacenaje de energía en forma de triglicéridos11. A lo largo de la gestación, pero fundamentalmente en el tercer trimestre, ambos, el TAM y el TAB, se acumulan influidos por el estado nutricional materno, desempeñando el TAM un papel esencial en la adaptación térmica del recién nacido al entorno extrauterino en el momento del parto10. Tras este, la diferenciación de adipocitos del TAB se acelera rápidamente y el TAM es sustituido, casi en su totalidad, por TAB. En este proceso, se identifican periodos de rápida acumulación de TAB, como ocurre en los primeros 18 meses de vida, en la media infancia y en la adolescencia, especialmente en las mujeres, preservando el TAB su capacidad adipogénica, esto es, para reclutar nuevos adipocitos desde preadipocitos, a lo largo de toda la vida adulta, aunque a un ritmo sustancialmente inferior12.
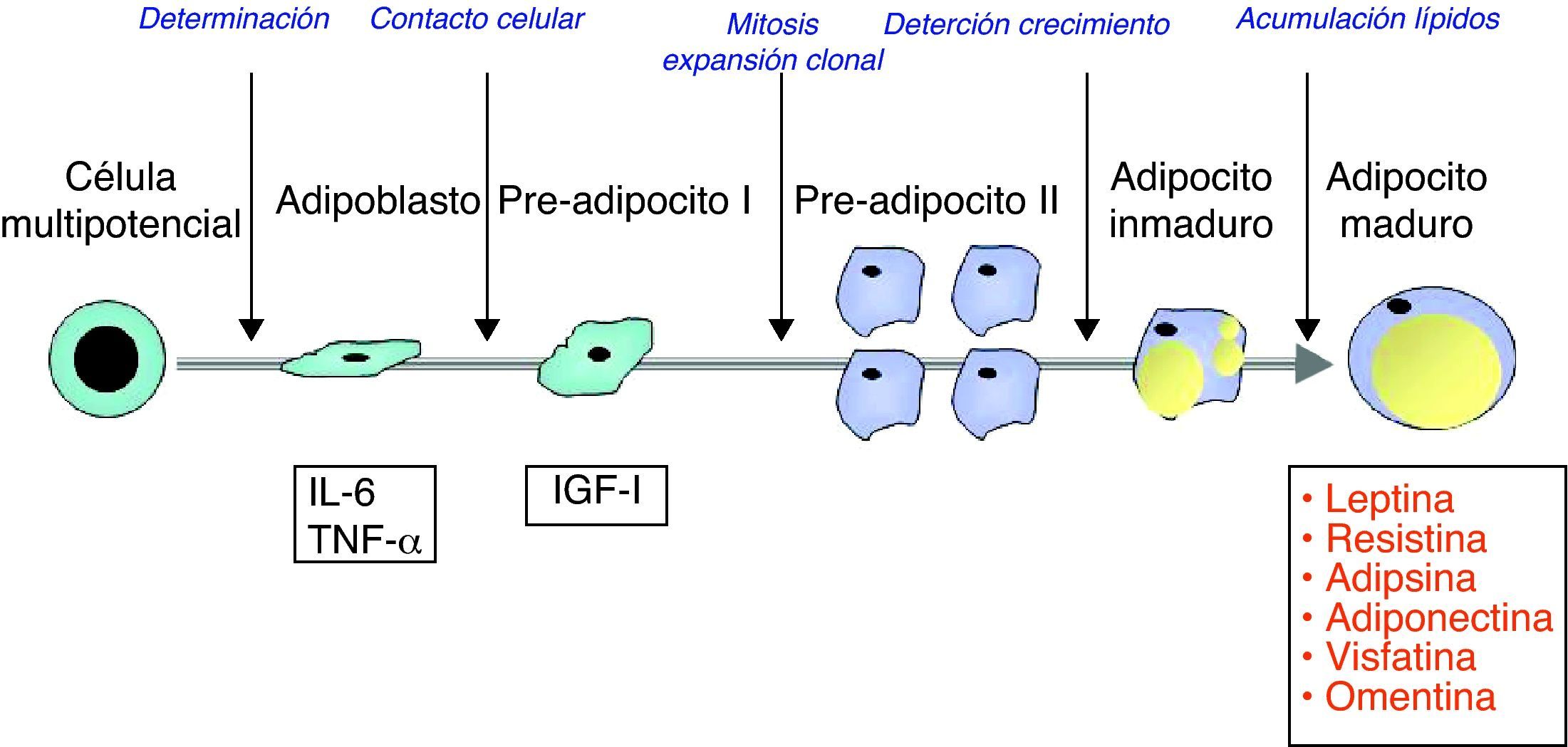
La secuencia de acontecimientos que conduce al desarrollo de adipocitos completamente diferenciados, a partir de progenitores pluripotenciales no diferenciados, recibe el nombre de adipogénesis y es distinta para el TAM y para el TAB. Durante la adipogénesis del TAB, los precursores embrionarios pluripotenciales dan lugar a células mesenquimatosas pluripotenciales que, tras su «reclutamiento» hacia la línea adipocitaria determinan, secuencialmente, la aparición de adipoblastos, preadipocitos tipo i y, tras su expansión clonal, preadipocitos tipo ii. Estos acumularán vacuolas lipídicas para la conformación de los adipocitos maduros. En esta secuencia de acontecimientos, la capacidad para sintetizar y secretar adipoquinas queda restringida de forma casi exclusiva, excepción hecha de algunas citoquinas proinflamatorias, a los adipocitos maduros13–15 (fig. 1).

Diferenciación adipogénica de las células mesenquimatosas pluripotenciales hasta el adipocito maduro. Nótese que la secreción de adipoquinas queda restringida, casi exclusivamente, al adipocito maduro. IGF-I: factor de crecimiento relacionado con la insulina número 1; IL-6: interleuquina 6; TNF-α: factor de necrosis tumoral alfa.
Como hemos referido con anterioridad, la matriz estromal del TA contiene múltiples subtipos celulares, principalmente derivados del SMF. Estas células, mayoritariamente monocitos y macrófagos, poseen la capacidad de secretar citoquinas (principalmente proinflamatorias) que, en combinación con las producidas por el adipocito, han terminado por conferir al TAB la catalogación como un auténtico órgano endocrino6. Este concepto fue reforzado posteriormente tras la demostración de la existencia de una comunicación efectiva bidireccional entre los adipocitos y el sistema nervioso central (SNC), particularmente el hipotálamo, así como con la hipófisis. De hecho, los adipocitos expresan receptores para las catecolaminas y para la mayoría de péptidos producidos por el hipotálamo y por la hipófisis6,16 (tabla 1).
Receptores hormonales expresados en el adipocito
| Receptores de catecolaminas | α1, α2 |
| β1, β2, β3 | |
| Receptores hormonales | |
| Membrana | Insulina, glucagón |
| GH, TSH, ACTH, CRH, prolactina | |
| Oxitocina | |
| Leptina, adiponectina | |
| Angiotensina II | |
| Nucleares | Hormonas tiroideas |
| Glucocorticoides | |
| Andrógenos, estrógenos, progesterona | |
| Vitamina D | |
| Receptores de citoquinas | Interleuquina 6 |
| Factor de necrosis tumoral alfa | |
ACTH: hormona corticotropa; CRH: péptido liberador de ACTH; GH: hormona de crecimiento; TSH: hormona estimuladora del tiroides.
Schäffler et al.17 propusieron el término «adipotropinas» para denominar al conjunto de hormonas y factores, producidos en el hipotálamo o la hipófisis, con efectos conocidos sobre los adipocitos16. Entre ellos, la hormona de crecimiento ejerce un efecto determinante en la acumulación y distribución del TA, así como en la regulación metabólica del mismo, estimulando la síntesis proteica y la lipólisis y antagonizando directamente la acción de la insulina18.
Modificaciones del tejido adiposo inducidas por la obesidad. Importancia de la edadEl desarrollo de obesidad determina la instauración de cambios histológicos, metabólicos y endocrinos en el TAB19–23. Estos cambios vienen determinados por distintos factores: a) la capacidad del TAB para el reclutamiento de nuevos adipocitos desde los preadipocitos, una vez que los adipocitos preexistentes han alcanzado un tamaño «crítico» mediante la modificación de las secreciones paracrinas de estos; en este proceso, denominado «hipótesis del tamaño crítico», la hipertrofia adipocitaria desempeña un papel esencial19; b) la capacidad del TAB para producir proteínas de quimioatracción (quimioquinas), que determinan un incremento de los subtipos proinflamatorios de monocitos y macrófagos20,24,25, con la consiguiente modificación del componente estromal de secreción de citoquinas22, y c) el cambio en el patrón de secreción endocrina de adipoquinas de los adipocitos hipertróficos, en comparación con el de aquellos más pequeños23,26.
La importancia relativa de cada uno de estos factores sobre las modificaciones ejercidas por la obesidad sobre el TAB varía a lo largo de las distintas etapas del desarrollo en el ser humano. Los adultos, tanto con normopeso como afectados de obesidad, mantienen una población adipocitaria, sobre la base de unas tasas proporcionadas de adipogénesis y apoptosis adipocitaria. Por el contrario, los niños y adolescentes incrementan, paulatinamente, el número de adipocitos de su TAB, con una tasa de proliferación superior en los pacientes obesos en comparación con los sujetos delgados27. Esto suscita la evidencia de que la obesidad de instauración precoz induce una tasa de reclutamiento de preadipocitos más acelerada, que determina un incremento de la población celular adipocitaria. Esta situación permitiría, al menos durante un tiempo, la existencia de un menor grado de hipertrofia adipocitaria, evitando o, al menos, atenuando la afectación del patrón de secreción de adipoquinas durante la infancia pero incrementando, por el contrario, el riesgo de obesidad severa y de desarrollo de comorbilidades en etapas posteriores de la vida2,27,28.
Este conjunto de acontecimientos remeda el concepto, ya antiguo, de la existencia de un modelo de «obesidad hiperplásica» en los niños, con un incremento del número de adipocitos, aunque con un tamaño normal, frente a un modelo de «obesidad hipertrófica» en el adulto, con un incremento del volumen de los adipocitos preexistentes. El contraste entre las características del TAB en ambos modelos de obesidad influye, de forma determinante, en la dinámica de secreción de adipoquinas a lo largo de las distintas etapas de la vida.
Control de la homeostasia energética: la leptina como pieza claveLa demostración de la implicación activa del TA en el control de la homeostasia energética aconteció con la clonación del gen de la leptina humana29 y la de su receptor específico30, describiéndose la vía de señalización leptina-proopiomelanocortina (POMC) como la fuente principal de información del SNC respecto a los depósitos energéticos del TAB en forma de triglicéridos. Dicha información se demostró, posteriormente, esencial para la regulación del crecimiento, el metabolismo y la reproducción.
La leptina es un polipéptido de 16 kDa producido, fundamentalmente, por los adipocitos maduros, aunque también por otros tejidos31. En el ser humano32,33 y en otros mamíferos34 la leptina actúa más como una señal de «adiposidad» que como una señal de saciedad, correlacionándose sus niveles séricos con el contenido graso corporal y fluctuando en relación con los cambios en el mismo y con su contenido en triglicéridos, aunque con una gran variabilidad interindividual para un mismo IMC32,33. La leptina circula en el torrente sanguíneo tanto libre como unida a proteínas, fundamentalmente a la isoforma soluble de su receptor específico, mostrando moderadas fluctuaciones siguiendo un ritmo circadiano, con mayores niveles nocturnos31.
El receptor de leptina (R-LEP) pertenece a la superfamilia de los receptores de citoquinas de clase i y se encuentra ubicuamente distribuido por la economía corporal permitiendo, mediante isoformas específicas, el transporte de la leptina a través de la barrera hematoencefálica (BHE). Su isoforma «larga» (la única capaz de activar sus vías de señalización) es particularmente abundante en el hipotálamo. Como hemos mencionado con anterioridad, existe una isoforma soluble de este receptor, que se une a la leptina de forma isomolecular, regulando su biodisponibilidad y su acción. Los niveles circulantes de esta isoforma del R-LEP pueden ser cuantificados y varían, de forma especular a los de leptina, bajo la influencia de los cambios en el contenido graso corporal31,35.
La leptina modula la actividad de múltiples poblaciones neuronales en el SNC, particularmente en el hipotálamo y en el tronco del encéfalo36. Los núcleos hipotalámicos estrechamente relacionados con la regulación de la ingesta incluyen: núcleos arcuato (NA), paraventricular (NPV) y dorsomedial (NDM), así como el hipotálamo ventromedial (HVM) y lateral (HL)37.
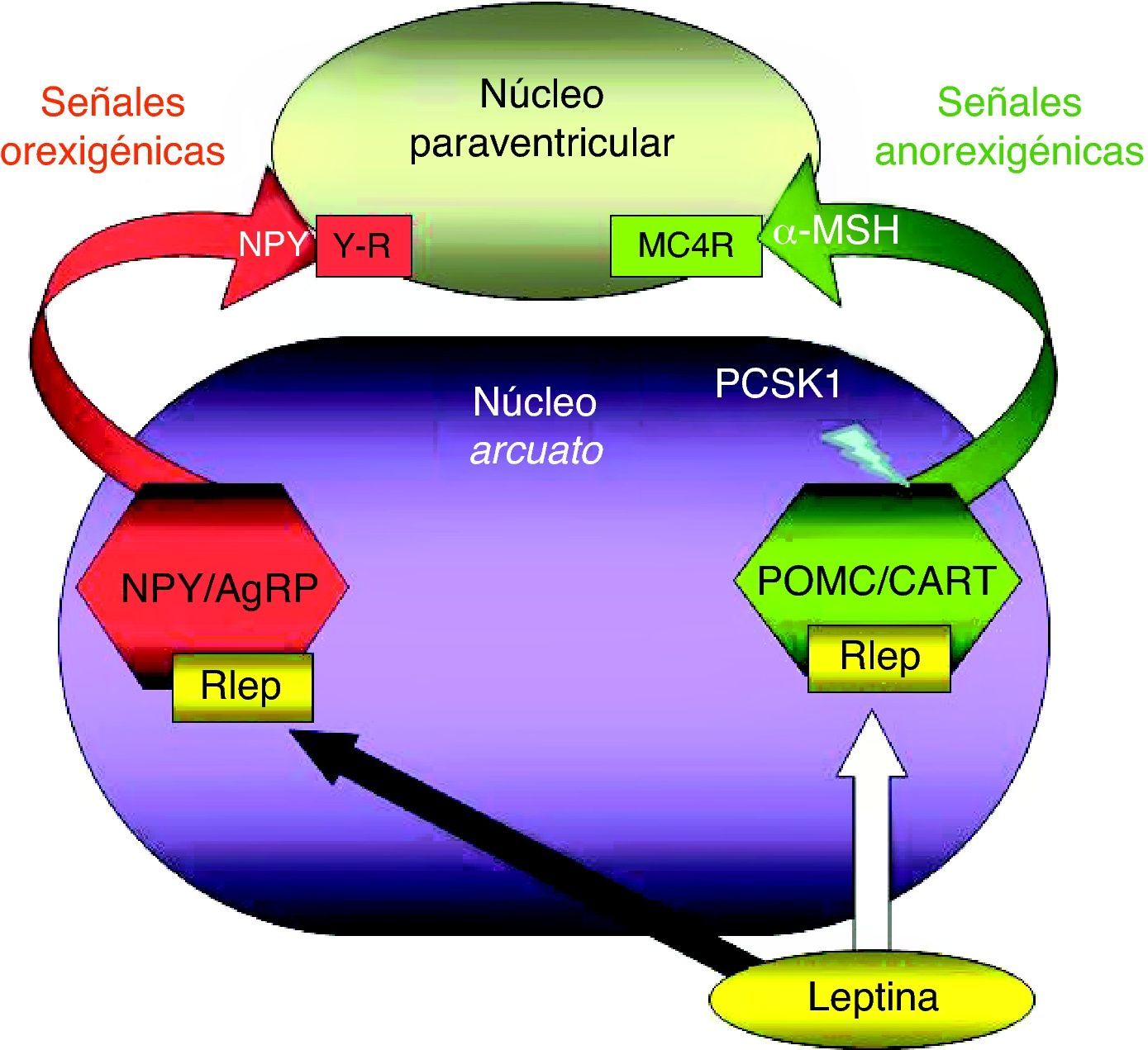
El NA contiene 2 poblaciones neuronales fundamentales para el equilibrio energético. Una de ellas estimula la ingesta mediante la liberación del neuropéptido Y (NPY) y del agouti-related peptide (AgRP)38. La segunda población, productora de POMC y del «transcrito regulado por cocaína y anfetamina» (CART), genera estímulos de saciedad al tiempo que inhibe los estímulos orexigénicos39. Ambas poblaciones neuronales expresan R-LEP y constituyen sitios de acción directos para la leptina, que estimula a las neuronas productoras de POMC/CART, al tiempo que inhibe a las generadoras de NPY/AgRP ejerciendo, por lo tanto, su función esencial como señal de suficiencia energética32,33 (fig. 2). Sin embargo, el incremento de los niveles séricos de leptina libre observado en la obesidad, resultante del aumento de secreción de leptina y de la disminución de su receptor soluble, no se reproduce en el líquido cefalorraquídeo ni, por tanto, en el hipotálamo. Este estado se ha descrito como «resistencia a la leptina» y parece ser consecuencia de la saturación de su transporte a través de la BHE, sumada a una señalización defectuosa de la misma en el hipotálamo. Esta situación, observada en la mayor parte de los pacientes afectados de obesidad, en cualquier edad, parece reversible, al menos de forma parcial, tras la reducción ponderal31,35.

Representación esquemática de la integración hipotalámica de los efectos anorexigénicos de la leptina en el núcleo arcuato hipotalámico. La flecha blanca indica su efecto estimulador sobre las neuronas productoras de POMC/CART, procesando la PCSK1 a la primera para producir α-MSH. La flecha negra indica su efecto inhibidor sobre las neuronas productoras de NPY/AgRP. α-MSH: fracción alfa de la hormona estimulante melanocítica; MC4R: receptor número 4 de MSH; NPY/AgRP: neuronas productoras de neuropéptido Y y del péptido relacionado con la proteína Agouti; PCSK1: convertasa de proproteínas tipo subtilisina kexina 1; POMC/CART: neuronas productoras de proopiomelanocortina y del transcrito relacionado con cocaína y anfetamina; RLEP: receptor de leptina.
La leptina completa su papel en la regulación de la homeostasia energética mediante un segundo mecanismo de acción, esta vez en los órganos periféricos. Así, estimula la cinasa dependiente de adenosín-monofosfato (AMP-K) en los miocitos, incrementando el gasto energético. Junto a este papel primordial en la regulación energética, la leptina está también implicada en la promoción del crecimiento, el metabolismo mineral óseo, la función inmunitaria, así como en la mitosis celular y en el desarrollo de distintos órganos, al tiempo que interviene en la regulación de la secreción hormonal de la mayor parte de los ejes hipotálamo-hipofisarios. La revisión detallada de todas las acciones ejercidas por la leptina excede las pretensiones de este artículo, para lo cual invitamos al lector a la consulta de otras referencias32–35,40.
Múltiples factores, que se ven influidos por el crecimiento y el desarrollo, modulan la síntesis de leptina, viéndose esta estimulada por la insulina, los estrógenos y los glucocorticoides, e inhibida por los andrógenos. Esta circunstancia enfatiza el interés especial que conlleva el estudio de la dinámica de la secreción y de las actividades de la leptina durante la infancia y, particularmente, en la obesidad infantil.
La edad gestacional y el peso en el momento del nacimiento determinan los niveles circulantes de leptina y su biodisponibilidad en el recién nacido (RN), mostrando los RN pretérminos y aquellos con peso bajo para su edad gestacional (PEG) niveles más bajos de leptina y más elevados de R-LEP en sangre de cordón que aquellos RN nacidos a término y con peso adecuado para su edad gestacional (PAEG), respectivamente41,42. De forma opuesta, los RN macrosómicos presentan una elevación de su leptina circulante43,44. El peso al nacimiento se ha señalado que podría constituir un factor predictivo de la posterior ganancia ponderal durante la infancia45, postulándose que los niveles de leptina al nacimiento serían un mejor indicador de la cantidad de TAB del RN que de su nivel de maduración46. Resulta interesante la observación de que existe una tendencia hacia una mayor concentración y biodisponibilidad de leptina en las niñas que en los niños en los partos a término, pero no en aquellas nacidas prematuramente41,45, lo cual parece indicar la existencia de un dimorfismo sexual de esta adipoquina ya desde edades tempranas de la vida.
Los niveles de leptina aumentan, de forma significativa, a lo largo del desarrollo puberal en las niñas y descienden, por el contrario, en la fase final de la pubertad en los varones. Por su parte, los niveles de R-LEP circulante disminuyen en ambos sexos tras el inicio de la pubertad, ocasionando un incremento del índice de leptina libre a lo largo del desarrollo puberal, más acentuado en las niñas47,48. Esto ha sido interpretado en clave de una señal del TA al SNC indicando la idoneidad de las condiciones de suficiencia energética para el desarrollo sexual y el establecimiento de la función reproductora, sobre todo en el sexo femenino48,49. Este cociente leptina/R-LEP, máximo en mujeres adultas, es el resultado de los cambios en el contenido graso corporal y en la producción de esteroides sexuales, ya que el IMC parece el mejor predictor de los niveles séricos de leptina libre50,51. Así, los esteroides sexuales ejercen un doble efecto sobre la síntesis de leptina, regulando la cantidad y distribución de los depósitos de TAB y, además, modulando directamente el control transcripcional de la leptina de forma positiva por los estrógenos y negativa por la testosterona52.
El impacto del IMC y del contenido graso corporal sobre los niveles circulantes y la biodisponibilidad de leptina es máximo en situaciones de malnutrición51. Se han observado niveles significativamente elevados de leptina y reducidos de R-LEP en niños obesos28,47,53,54 y la imagen opuesta se observa en situaciones de malnutrición, como la anorexia nerviosa47. Estos cambios opuestos de leptina y R-LEP en la obesidad determinan la afectación de su señalización, como consecuencia, al menos en parte, de la saturación del R-LEP55. Además, existe una influencia bidireccional entre leptina e insulina, de modo que la hiperinsulinemia exacerba la producción de leptina56, al tiempo que la elevación del índice de leptina libre intensifica la resistencia periférica a la acción de la insulina57.
El papel ejercido por el contenido graso corporal sobre los niveles circulantes de leptina y su biodisponibilidad es reforzado por el descenso de los primeros y el incremento de los segundos evidenciados tras una reducción ponderal en niños y adolescentes28,47,53. Sin embargo, de forma opuesta al estado de «resistencia o insensibilidad a leptina», previamente mencionado en los adultos, estudios recientes apuntan que unos niveles elevados de leptina en situación basal en niños obesos pueden verse sucedidos de un gran descenso de los mismos tras la reducción ponderal, pudiendo emplearse como eventuales predictores de la reducción de contenido graso corporal a corto y largo plazo. Así, no tienen por qué, necesariamente, reflejar la existencia de «resistencia a la leptina» en edades tempranas58.
La confirmación definitiva de la importancia de la leptina en el balance energético y sobre el desarrollo puberal derivó de la constatación de casos humanos de deficiencia congénita de leptina59 y deficiencia congénita de R-LEP60, de base genética. Estos sujetos presentan una obesidad extremadamente grave, de inicio precoz y ausencia de desarrollo puberal, que puede resolverse tras la administración de leptina biosintética61.
La implicación de otras adipoquinas en el control de la homeostasia energética es mucho menos evidente, pero no puede ser descartada. Así, los receptores específicos de adiponectina se encuentran ampliamente distribuidos por el cerebro y la inyección intracerebroventricular (icv) de la misma determina una reducción del peso corporal debido a un incremento del gasto energético62, como también la de interleuquina 6 (IL-6)63, al tiempo que se ha comunicado la inhibición de la ingesta alimentaria tras la administración icv de resistina62,64. Sin embargo, la implicación fisiopatológica de estas adipoquinas en el control de la homeostasis energética dista de ser bien conocida.
Sensibilidad a la insulina: papel de adiponectina, visfatina, vaspina y omentinaLa resistencia periférica a la acción de la insulina, o resistencia a insulina (RI), fue postulada por Gerald Reaven como la base fisiopatológica del resto de alteraciones metabólicas asociadas a la obesidad y denominadas síndrome metabólico (SM) o síndrome X65.
El papel desempeñado por la cantidad y, de forma más importante, la distribución del TAB acumulado, en la génesis de la RI ha sido estudiado profusamente, resaltando la especial implicación del TAB visceral en este proceso66. Uno de los determinantes mayores del desarrollo de RI asociada a obesidad es el cambio en el patrón de secreción de adipoquinas por parte del TA de los pacientes obesos, especialmente niños. Entre las adipoquinas implicadas en la sensibilidad a la acción de la insulina y la génesis de RI se encuentran las citoquinas proinflamatorias, incluyendo la resistina, pero muy particularmente la adiponectina y las más recientemente caracterizadas visfatina, vaspina y omentina.
AdiponectinaLa adiponectina es un péptido de 30 kDa con un dominio similar al colágeno que permite la formación de estructuras secundarias y terciarias. Está producida exclusivamente por los adipocitos maduros, con mayor síntesis por parte de aquellos localizados en el TAB subcutáneo respecto a los del TAB visceral. En el torrente circulatorio, los monómeros de adiponectina se unen formando homotrímeros que, a su vez, pueden conformar estructuras más complejas, como hexámeros de, aproximadamente, 180 kDa (bajo peso molecular [LMW]) y polímeros (16-18 monómeros) de 400-600 kDa (alto peso molecular [HMW]). La forma HMW de la adiponectina es la más abundante en suero y se ha postulado como la determinante de las acciones metabólicas fundamentales de la adiponectina. Por ejemplo, el grado de sensibilidad a la acción periférica de la insulina parece correlacionar mejor con el cociente entre adiponectina HMW/total (SA) que con los niveles totales (T) de adiponectina circulante6,31,67–70.
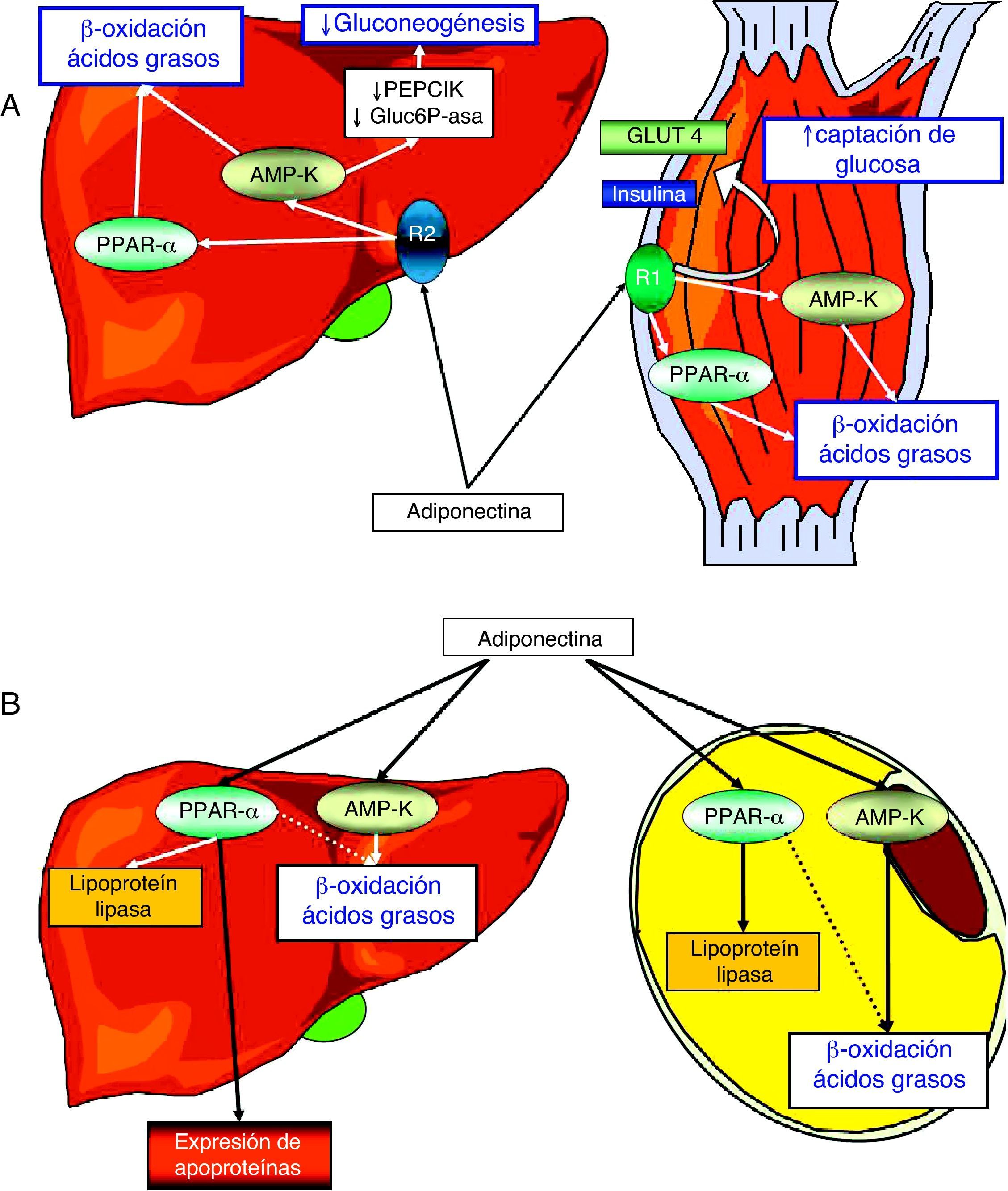
Posee 2 receptores específicos, adipoR1 y R2, distribuidos de forma ubicua por el organismo, pero expresados de forma mayoritaria en el músculo y en el hígado, respectivamente. La activación del adipoR1 muscular determina la estimulación de la AMP-K (como también provoca la leptina por medio de su receptor), lo que induce la proliferación del PPAR-α (peroxisome proliferator activated receptor alpha) y, como consecuencia, la expresión de enzimas implicadas en el catabolismo de los ácidos grasos y en la captación de glucosa. Además, estimula la expresión y transporte hacia la membrana celular del miocito, del transportador de glucosa número 4 (Glut-4, al tiempo que modula directamente la señalización del receptor de insulina. Por su parte, la estimulación del receptor adipoR2 hepático y la subsiguiente activación de la AMP-K inhiben la gluconeogénesis, mediante la modulación de la actividad de la glucosa-6-fosfatasa y de la fosfo-enol-piruvato descarboxilasa9. El TAB también recibe la acción de la adiponectina, favoreciendo en él la diferenciación adipocitaria, la captación de glucosa, la oxidación de ácidos grasos y la actividad lipoproteín-lipasa71,72. En conjunto, los efectos de la adiponectina sobre el músculo, el hígado y el TAB resultan en un incremento de la sensibilidad a la captación periférica de glucosa inducida por insulina y en la promoción de la oxidación de ácidos grasos y de un perfil de apoproteínas beneficioso6,31,67,70,73,74 (fig. 3 A y B).
 y sobre el metabolismo lipídico en el hígado y los adipocitos (B). La estimulación del receptor adipoR1 muscular determina la activación de la quinasa dependiente de AMP (AMP-K) e induce la expresión de PPAR-α (peroxisome proliferator activated receptor alpha) y, como consecuencia, la expresión de enzimas implicadas en la captación de glucosa y en el catabolismo de los ácidos grasos. Además, la adiponectina estimula la expresión y externalización del transportador de glucosa número 4 (Glut-4) en el miocito, al tiempo que modula directamente la actividad del receptor de insulina. La activación del receptor adipoR2 y de la AMP-K hepáticos inhibe la gluconeogénesis mediante la modulación de la actividad de la glucosa-6-fosfatasa (Gluc6P-asa) y de la fosfo-enol-piruvato-descarboxilasa (PEPCK), estimulando la actividad de la lipoproteín-lipasa y modulando la expresión génica de las apoproteínas. Finalmente, la adiponectina favorece la captación de glucosa estimulada por insulina, la oxidación de ácidos grasos y la actividad de la lipoproteín-lipasa en los adipocitos del tejido adiposo blanco.")
Representación esquemática de las principales acciones metabólicas de la adiponectina sobre el metabolismo de los hidratos de carbono en el hígado y el músculo (A) y sobre el metabolismo lipídico en el hígado y los adipocitos (B). La estimulación del receptor adipoR1 muscular determina la activación de la quinasa dependiente de AMP (AMP-K) e induce la expresión de PPAR-α (peroxisome proliferator activated receptor alpha) y, como consecuencia, la expresión de enzimas implicadas en la captación de glucosa y en el catabolismo de los ácidos grasos. Además, la adiponectina estimula la expresión y externalización del transportador de glucosa número 4 (Glut-4) en el miocito, al tiempo que modula directamente la actividad del receptor de insulina. La activación del receptor adipoR2 y de la AMP-K hepáticos inhibe la gluconeogénesis mediante la modulación de la actividad de la glucosa-6-fosfatasa (Gluc6P-asa) y de la fosfo-enol-piruvato-descarboxilasa (PEPCK), estimulando la actividad de la lipoproteín-lipasa y modulando la expresión génica de las apoproteínas. Finalmente, la adiponectina favorece la captación de glucosa estimulada por insulina, la oxidación de ácidos grasos y la actividad de la lipoproteín-lipasa en los adipocitos del tejido adiposo blanco.
Existe evidencia de la implicación de la adiponectina en la regulación del metabolismo de los hidratos de carbono, que incluye la disminución de los niveles séricos de esta adipoquina en pacientes afectados de diabetes mellitus tipo 2 (DM2), de forma independiente de su grado de adiposidad. La deficiencia genética de adiponectina se asocia al desarrollo de RI, mientras que su administración a modelos experimentales aumenta la sensibilidad a la acción de la insulina. Esta parece, además, regular negativamente la expresión de adiponectina. Sin embargo, aunque la influencia de la adiponectina sobre el metabolismo de los hidratos de carbono parece evidente, los datos disponibles actualmente no permiten establecer una relación inequívoca entre los niveles de adiponectina y el grado de sensibilidad a la insulina. De hecho, algunos autores señalan que esta adipoquina podría ser únicamente un marcador, más que un componente activamente implicado en la modulación de la sensibilidad a la insulina.
Al contrario que lo descrito anteriormente para la leptina, los niveles séricos de insulina se correlacionan de forma inversa con el contenido corporal de TAB, observándose niveles disminuidos de adiponectina en adultos obesos75. Sin embargo, existe controversia respecto a la influencia que, sobre la adiponectina, ejerce la distribución del TAB corporal entre sus depósitos, subcutáneo y visceral68. De forma interesante, la correlación inversa existente entre el contenido graso corporal y los niveles circulantes de adiponectina no está presente a lo largo de toda la vida extrauterina, al tiempo que se ve influida, también, por los cambios estructurales y funcionales ejercidos sobre el TA por determinados tratamientos farmacológicos (como las tiazolidinedionas)72.
En los RN, al igual que ocurre con la leptina, existe una correlación positiva entre los niveles de adiponectina, la edad gestacional y el peso en el momento del nacimiento41,76, opuesta a lo observado en adultos, con niveles más altos en los RN de sexo femenino41,77. En el momento del nacimiento, los niveles de adiponectina sérica duplican o incluso triplican los del adulto, con un posterior descenso y desaparición de su correlación positiva con el peso corporal hacia los 2 años de vida extrauterina, coincidiendo con uno de los periodos de rápida adipogénesis68. Su dimorfismo sexual desaparece durante el periodo prepuberal48,68, pero vuelve a aparecer, de forma más intensa, a partir de la media pubertad, con un descenso en los niveles exhibidos por los varones, probablemente como consecuencia del efecto inhibidor que, sobre la síntesis de adiponectina, ejercen los andrógenos y de la mayor acumulación de TA visceral en el sexo masculino48,68,70,74,78.
En adolescentes obesos ya se observa la correlación negativa entre contenido graso corporal, RI y niveles circulantes de adiponectina, descrita en los adultos68,79,80. Sin embargo, mientras la asociación negativa existente entre RI y adiponectina sí que se observa ya en niños prepuberales69, existen resultados contradictorios respecto a la correlación existente entre adiponectina e IMC en este periodo vital. Algunos estudios comunican la existencia de una correlación negativa80 o la ausencia de correlación28 entre ambos. Así como niveles de adiponectina total en niños obesos tanto reducidos68,81 como similares28 a lo de sus coetáneos delgados. Como comentamos con anterioridad, esto puede ser debido a la mayor capacidad del tejido para el reclutamiento de nuevos adipocitos desde los preadipocitos en estas edades tempranas de la vida, lo que podría permitir limitar la hipertrofia de los adipocitos preexistentes y, por lo tanto, el mantenimiento de una población adipocitaria de «tamaño adecuado», sin afectarse su capacidad para la secreción de adiponectina, al menos durante algún tiempo26,27. De cualquier modo, el estudio específico de los complejos de HMW-adiponectina ha mostrado que, incluso en estas edades, la síntesis, secreción y posterior procesamiento postraduccional de la adiponectina se ve afectado, asociando un incremento de RI28. De forma interesante y análoga a las observaciones en adultos, la reducción ponderal se asocia a un incremento de los niveles circulantes de adiponectina a cualquier edad, probablemente debido a una «optimización» del tamaño de toda la población adipocitaria del niño y, secundariamente, de su patrón secretor de adipoquinas28,68.
La adiponectina producida por los adipocitos localizados en el TA perivascular ejercen un efecto beneficioso inhibiendo el desarrollo de arteriosclerosis mediante diversos mecanismos como son: la modulación de la reactividad vascular dependiente del endotelio, inhibición de la expresión de moléculas de adhesión e inflamación, inhibición de la formación de células espumosas e inhibición de las metaloproteasas, responsables de la fractura de la placa de ateroma67.
Se ha indicado que la hipoadiponectinemia asociada a la obesidad podría estar implicada en la generación del estado de inflamación de baja intensidad que acompaña a la misma, sobre la base de su potencial efecto antiinflamatorio70 y su asociación a niveles elevados de proteína C reactiva (PCR), IL-6 y TNF-α74. Contrariamente, la evidencia de un eventual papel protector de la adiponectina frente al desarrollo de esteatohepatitis no alcohólica en niños es limitada68, si bien esto ha sido demostrado en el caso de los pacientes adultos70.
Visfatina/factor estimulador de colonias pre-B-cell/nicotinamida-pfosforibosiltransferasaLa visfatina o NAMPT es un péptido de 52 kDa cuyo nombre deriva de la asunción inicial de que su principal fuente de producción era el TAB de localización visceral. Se le ha postulado un eventual efecto hipoglucemiante, al tiempo que los estudios in vitro han mostrado que es capaz de inducir la fosforilación del receptor de insulina y de sus sustratos relacionados (IRS) 1 y 282. Esta caracterización inicial hizo prever un papel esencial de esta adipoquina en el vínculo entre la obesidad de predominio visceral y el desarrollo de RI y DM2, reforzado posteriormente tras la demostración de su implicación en la secreción de insulina83. Sin embargo, este péptido se encuentra en una concentración muy baja en el torrente circulatorio, y los estudios posteriores que han intentado caracterizar su relación con el metabolismo de los hidratos de carbono muestran resultados contradictorios84. Es más, estudios recientes demuestran que la fuente fundamental de la visfatina no es el TA visceral, sino los leucocitos, lo cual señala que sus acciones estarían mayoritariamente relacionadas con la inflamación y no con la regulación metabólica85. Las múltiples incertidumbres existentes respecto a la trascendencia fisiopatológica y a las acciones de esta adipoquina han generado un cierto grado de escepticismo respecto a su verdadera importancia funcional86.
En adultos, se ha descrito la existencia de una correlación positiva entre los niveles séricos de visfatina y el contenido graso corporal, así como un descenso de los mismos tras la reducción ponderal87. Sin embargo, la evidencia disponible respecto a una eventual mayor producción por parte del TA visceral es inconsistente83,88. Algunos autores señalan que esta adipoquina podría ser, únicamente, un marcador de la cantidad total de TAB corporal. Más aún, el efecto de la reducción ponderal sobre los niveles séricos de visfatina en adultos permanece en discusión debido a la existencia de resultados contradictorios, mostrando tanto su disminución89 como su aumento90 tras intervenciones de cirugía bariátrica.
Los niveles de visfatina durante el periodo fetal son elevados, probablemente debido a la transferencia materna y a la producción placentaria91, habiéndose comunicado una correlación positiva de los mismos con el peso corporal, pero tanto en RN pretérminos como a término91,92. Esto concuerda con la mayoría93,94, aunque no con todos95, los estudios en RN macrosómicos. Igualmente, el efecto de la restricción del crecimiento intrauterino sobre los niveles de visfatina es controvertido, y se han comunicado, en RN PEG, niveles de visfatina similares96 o superiores91 a los de los RN con PAEG. Una explicación plausible para estas discrepancias podría ser la existencia de dimorfismo sexual, con niveles superiores comunicados en las niñas97.
Los estudios de los niveles de visfatina en niños delgados son escasos85,98 y la mayor parte provienen del estudio de grupos de controles empleados en la comparación con niños95,99. Estos estudios muestran una correlación positiva entre visfatina e IMC, incluso en niños delgados98, sin demostración de dimorfismo sexual en la infancia o adolescencia, ni variaciones en sus niveles como consecuencia de la progresión de la misma en ninguno de los 2 sexos85. Además, no se ha observado correlación de los niveles de visfatina con el crecimiento, ni con sus marcadores (IGF-I), ni con los esteroides sexuales85,98,99.
Se ha descrito la existencia de mayores concentraciones séricas de visfatina en niños obesos que en controles, así como una correlación positiva de estos con el IMC y con los marcadores surrogados del contenido graso corporal (leptina y R-LEP), en varias series de niños y adolescentes85,100–102, así como específicamente en el periodo prepuberal103. En estas edades, la correlación de los niveles de visfatina con los indicadores de adiposidad abdominal (perímetro de la cintura) es incierta, lo cual parece indicar que dependen, fundamentalmente, de la cantidad total de grasa corporal y no de su distribución100,103. La reducción ponderal determina un descenso inicial de visfatina103,104, que permanece estable cuando esta se intensifica103.
Respecto al papel fisiopatológico de la visfatina en la obesidad infantil, se ha descrito que la relación existente entre sus niveles y la alteración del metabolismo de los hidratos de carbono está influida parcial, pero no completamente, por el IMC. Así, el descenso de los niveles de visfatina como consecuencia de la reducción ponderal, paralelo a la disminución en el índice HOMA de resistencia a la insulina, no está mediado exclusivamente por la disminución del contenido graso corporal, sino también por la mejora en la sensibilidad a insulina. De hecho, la insulina parece disminuir, directamente, la síntesis y secreción de visfatina105. Aunque la mayor parte de los estudios muestran que la correlación existente entre visfatina e índice HOMA desaparece tras controlar el efecto del IMC102,103, la disminución de los niveles de visfatina a lo largo de un test de tolerancia oral a la glucosa (TTOG) parece estar vehiculada por la insulina85,105, incrementándose este efecto por el efecto sensibilizante a la insulina de la reducción ponderal103. Este cuerpo de evidencia señala que, ya que los niños habitualmente no presentan alteraciones mayores del metabolismo de los hidratos de carbono, los niveles elevados de insulina en niños obesos podrían constituir un mecanismo de compensación dirigido a mantener la homeostasia metabólica a largo plazo, que se normalizaría tras la reducción ponderal. Por el contrario, el brusco incremento de la insulinemia que acontece en el TTOG determinaría una disminución de este perfil favorable a la sensibilidad a la insulina.
También se ha señalado la existencia de una relación entre la visfatina y el metabolismo lipídico, con el hallazgo de correlación entre los niveles de ácido ribonucleico mensajero de visfatina, tanto circulante como tisular, y los niveles de colesterol circulante en adultos delgados y obesos106. En los niños, la correlación entre visfatina y colesterol circulante desaparece tras controlar el efecto ejercido por el IMC99,103, por lo que se considera que puede ser un epifenómeno asociado a los cambios en este.
Finalmente, es conocido que la visfatina está producida, fundamentalmente, por los leucocitos circulantes85 y la fracción estromal del TAB bajo la influencia de estímulos proinflamatorios107,108. Además, sus niveles se correlacionan positivamente con otras citoquinas proinflamatorias como IL-6103,109, TNF-α98 o resistina103, independientemente del IMC. Esto enfatiza su contribución a la generación del estado inflamatorio de baja intensidad asociado a la obesidad.
VaspinaLa vaspina (visceral adipose-tissue derived serine protease inhibitor) es un péptido de 515 aminoácidos perteneciente a la familia de las serpinas (serine protease inhibitor)110. En adultos humanos, su expresión se ha comprobado tanto en el TAB subcutáneo, como en el visceral, postulándose una regulación de la misma ejercida por el contenido graso corporal y por el estatus de sensibilidad a insulina111,112. La administración de vaspina reduce la RI, mejorando la tolerancia a glucosa y la ingesta en ratones obesos113. Sin embargo, su mecanismo de acción y sus dianas moleculares permanecen sin caracterizar110,112, por lo que su papel como sensibilizante endógeno a la acción de la insulina es muy debatido.
Los niveles séricos de vaspina son superiores en las mujeres adultas respecto a los hombres112,114 y expresan una variación diurna relacionada con la ingesta115. Sin embargo, existen datos contradictorios referentes a la relación entre los niveles de vaspina, el IMC y las alteraciones del metabolismo de los hidratos de carbono en el hombre116,117, así como respecto a la influencia de la reducción ponderal sobre sus niveles circulantes106,114.
La información disponible respecto a vaspina en niños es limitada. Los escasos estudios realizados en RN muestran que la vaspina es ya detectable en sangre de cordón y que no muestra relación con los niveles de insulina ni con el peso al nacimiento, sin existir diferencias entre RN PEG, con PAEG o macrosómicos94,118. El dimorfismo sexual observado en adultos se establece durante la pubertad en niños sanos, observándose niveles estables en los niños y progresivamente más altos en las niñas conforme progresa el desarrollo puberal, sin relación de los niveles de vaspina con el IMC119.
Dos estudios recientes comunican la existencia de niveles elevados de vaspina en niños y adolescentes obesos120 y un descenso de los mismos tras tratamiento121. Sin embargo, los resultados relativos a la relación entre vaspina y el índice HOMA o la insulinemia son conflictivos, comunicándose correlaciones tanto negativas119,121 como positivas120 con los mismos. En el periodo prepuberal, los niveles de vaspina no muestran diferencias entre sexos, ni entre niños con obesidad o normopeso, ni se modifican tras la reducción ponderal en los primeros103. Más aún, no se han podido establecer correlaciones significativas entre vaspina, IMC, HOMA o insulinemia en niños ni en adultos sanos117, aunque sí en pacientes con DM-2. Esto sugiere, al igual que hemos referido para la visfatina, que la ausencia de alteraciones mayores del metabolismo de los hidratos de carbono en las cohortes más jóvenes estudiadas puede ser la causa de esta ausencia de correlaciones.
La ingesta de glucosa determina una disminución de los niveles de vaspina en niños y adolescentes obesos e hiperinsulinémicos103,119, así como en modelos animales122, habiéndose observado un descenso en la vaspina sérica en adultos sanos tras la ingesta115. Llama la atención que esta disminución de vaspina inducida por glucosa no se observó en niños y adolescentes normoinsulinémicos119, ni en niños prepuberales tras la reducción ponderal103, si bien en estos últimos sí que existía correlación entre las áreas bajo la curva de vaspina e insulina en el TTOG realizado tras la reducción ponderal, pero no en el realizado antes de la misma103. Estas observaciones avalan la hipótesis de un sistema regulatorio complejo que implica una coordinación entre el contenido graso corporal y el estatus de sensibilidad a la insulina como principales reguladores de la secreción de vaspina.
OmentinaLa omentina 1, también llamada intelectina123, es una adipoquina de 313 aminoácidos, producida mayoritariamente por la matriz estromal del TAB visceral124. Su análogo, omentina 2, comparte con ella un 83% de analogía en su composición aminoacídica. Se han postulado 3 potenciales acciones para la omentina 1: a) incremento de la captación de glucosa mediada por insulina124,125; b) inhibición de la inflamación y angiogénesis inducida por la PCR125, y c) vasodilatación arterial. En conjunto, se le atribuye un papel protector frente al SM.
En humanos adultos, sus niveles plasmáticos y expresión génica aparecen disminuidos en la obesidad126 y en situaciones de RI127. En niños y adolescentes, la información disponible es mínima, aunque se sabe que la omentina 1 está presente ya en vida fetal, sin correlacionarse al nacimiento ni con el peso ni con los niveles de insulinemia128. En niños prepuberales sanos (n=161) se ha comunicado la asociación de niveles elevados de omentina 1 con un peor perfil metabólico (mayor índice HOMA, triglicéridos y tensión arterial), sin aparente influencia del sexo, de la edad o del IMC129.
Otras adipoquinas con implicación en el metabolismo de los hidratos de carbonoSe ha propuesto que muchas otras adipoquinas pueden estar implicadas en la regulación del metabolismo de los hidratos de carbono. Entre ellas se cuenta la apelina, para cuya asociación con el IMC o la RI en niños no existe evidencia130, existiendo datos contradictorios en adolescentes131. También se ha sugerido un papel de la proteína fijadora de retinol número 4 (RBP4), con resultados contradictorios en cuanto a su implicación en el metabolismo hidrocarbonado en la infancia y adolescencia; así como de la quimerina, sin estudios disponibles en niños132.
Estado proinflamatorio de baja intensidad: adipoquinas proinflamatoriasEl estado proinflamatorio descrito en relación con la obesidad es consecuencia de múltiples factores anteriormente descritos (cambios morfológicos y funcionales de los adipocitos y de las células del SMF contenidas en el estroma y modificación del patrón de secreción de adipoquinas)22,132,133. Ya hemos esbozado las acciones proinflamatorias (NAMPT/visfatina) y antiinflamatorias (adiponectina) de las adipoquinas clínicamente más relevantes y específicas del TA. Junto a ellas, cabe destacar otras citoquinas proinflamatorias que, si bien también se producen en el TA, su secreción se produce, mayoritariamente, por parte de las células del SMF de cualquier localización anatómica. Por este motivo, su especificidad y utilidad como marcadores de la actividad endocrina del TA queda muy limitada.
ResistinaEsencial en el desarrollo de la RI asociada a obesidad en modelos murinos, en el ser humano esta citoquina, perteneciente a la familia FIZZ (found in inflammatory zone) ejerce un efecto fundamentalmente proinflamatorio. Esta diferencia responde, sobre todo, a la limitada homología estructural entre especies (en torno al 60%) y, de forma más importante, a la diferencia en su fuente de producción principal: adipocitos en el ratón, células mononucleares en el ser humano134. De cualquier modo, recientes estudios de casos y controles en humanos han demostrado la existencia de un riesgo incrementado de desarrollo de DM2 en pacientes con niveles elevados de resistina135.
En RN, los niveles de resistina de los pretérmino son superiores a los de los RN a término41, lo cual podría estar relacionado con la mayor prevalencia de un entorno proinflamatorio en estos embarazos136. Esto está avalado por el hallazgo de correlaciones positivas entre los niveles de resistina y los de otras citoquinas proinflamatorias en sangre de cordón, así como por el descenso de los mismos tras la administración de corticoides antenatales41,137. Existen asimismo datos contradictorios referentes a la relación existente entre los niveles de resistina y el peso en el momento del nacimiento, sin haberse demostrado relación con los niveles de insulinemia41,118,137–139.
En niños sanos, los niveles séricos de resistina son superiores en niñas a lo largo del desarrollo puberal48,140–142, existiendo correlación de los mismos con los de estradiol141 y con el cociente leptina/R-LEP en niñas, pero no en el sexo masculino. En una gran serie que agrupaba niños con y sin obesidad, la resistina demostró ser un marcador poco robusto de obesidad o inflamación, sin hallarse correlación con los índices de RI143.
Una vez más, al comparar los niveles circulantes de resistina entre niños obesos y controles, los resultados son contradictorios, observándose niveles similares141,144 o superiores en los pacientes obesos28,145. Del mismo modo, su fluctuación tras la reducción ponderal144,146 o en respuesta a la actividad física146,147 son contradictorios, sin hallarse correlación entre los niveles de resistina y las mediciones directas del contenido graso corporal, ni con los índices de RI en niños obesos28.
Interleuquina 6 y factor de necrosis tumoral alfaLa IL-6 y el TNF-α son secretados, como la resistina, principalmente por las células del SMF de cualquier localización, aunque los adipocitos también sintetizan y secretan ambas, al tiempo que expresan sus receptores específicos. La producción de ambas citoquinas por los adipocitos es mayor en estados de obesidad6,148. El TNF-α actúa fundamentalmente de forma paracrina, circula en muy bajas concentraciones en suero y es de producción ubicua, en contraste con la IL-6, para la que se estima que aproximadamente un tercio de la que circula en suero procede del TAB (fundamentalmente visceral), habiéndose comunicado correlación de los niveles de esta con el IMC y los índices de RI6,148,149. De cualquier modo, incluso en pacientes con obesidad mórbida, los niveles séricos de IL-6 están próximos al límite superior de la normalidad, lo que limita su utilidad diagnóstica6, si bien recientemente se ha postulado la posibilidad de que sea uno de los marcadores más precoces del inicio de las alteraciones asociadas al desarrollo de obesidad en sujetos más jóvenes149.
Como ocurría con la resistina, los niveles de IL-6 son mayores en RN pretérmino que en RN a término41. Sin embargo, los niveles de IL-6 más bajos en RN PEG respecto a aquellos con PAEG podrían estar influidos por su menor contenido graso corporal41. Por el contrario, en el mismo estudio no se observaron cambios en los niveles de TNF-α en relación con la edad gestacional ni con el peso en el momento del nacimiento41. Esto puede verse influido, además de por la acción preferentemente paracrina del TNF-α, por la polarización del sistema inmunitario innato del RN hacia la IL-6 en detrimento del TNF-α150.
En niños sanos, los niveles de IL-6 no difieren entre sexos, disminuyendo tanto en niños como en niñas tras la instauración de la pubertad, con una correlación negativa con los esteroides sexuales, cuyo efecto inhibidor sobre la producción de IL-6 es conocido151,152. En cambio, los niveles de TNF-α no varían significativamente a lo largo de la pubertad, ni muestran diferencias entre sexos153.
Los niños obesos presentan niveles elevados de IL-6 y de TNF-α28, en comparación con niños delgados y con aquellos especialmente elevados en los pacientes con intolerancia a los hidratos de carbono154. Ambas citoquinas muestran una correlación positiva con el IMC, reduciéndose los niveles de IL-6 en un corto periodo de tiempo tras la reducción ponderal, mientras que los niveles de TNF-α precisan una reducción ponderal mantenida en el tiempo para experimentar dicho descenso28
Se han estudiado otras citoquinas proinflamatorias en la obesidad infantil, comunicándose la existencia de una correlación negativa entre el IMC y los niveles séricos de IL-10 (antagonista de las acciones de IL-6 y TNF-α) en niños155. También se ha comprobado que los niveles de IL-1β e IL-8 no muestran dimorfismo sexual a lo largo de la infancia o adolescencia en niños sanos, pero que la primera disminuye y la segunda aumenta en los estadios finales del desarrollo puberal28, aunque el significado de estos cambios y su eventual trascendencia en el desarrollo de obesidad son, actualmente, desconocidos.
Resumen y conclusionesEn este artículo especial hemos resumido el estado actual del conocimiento referente a la dinámica de secreción y a las acciones de las adipoquinas en la infancia, concentrándonos en sus papeles en el control de la homeostasia energética, regulación metabólica e inflamación. También hemos discutido las particularidades de las mismas en estas etapas del desarrollo, frente a los datos conocidos en el adulto, no siempre extrapolables, así como la influencia ejercida por la duración y el crecimiento durante el desarrollo intrauterino y, posteriormente, extrauterino, y las modificaciones establecidas por la instauración de la obesidad en etapas precoces de la vida.
Confiamos en que la lectura del mismo permita desprender la conclusión evidente de que en el caso de la obesidad, como en el de muchas otras enfermedades, los niños no son exclusivamente «adultos pequeños». En particular, en el caso de la obesidad, esta diferencia es particularmente importante debido a la extraordinaria plasticidad de su TA, que les confiere características diferenciales que, a su vez, evolucionan conforme progresa el crecimiento y el desarrollo puberal, con la influencia del sexo del niño. En consecuencia, la edad a la que se produce el establecimiento de la obesidad, así como la intensidad de la misma, influye en la estructura y función del TAB, constituyendo la secreción y las acciones de la adiponectina excelente ejemplo de ello.
Finalmente, la documentación de la presencia de la mayor parte de las adipoquinas durante la vida fetal, su papel en el equilibrio energético y su influencia sobre procesos fisiológicos como el crecimiento y la pubertad, deben desterrar la catalogación del TA como un órgano «pasivo». Así, teniendo en cuenta la existencia de diferencias funcionales entre los distintos depósitos de TAB sería prudente considerar la existencia de distintos «órganos adiposos» que establecen comunicación bidireccional con el SNC, con los diferentes ejes endocrinos y con toda la economía corporal, desempeñando un papel extraordinariamente activo en el mantenimiento de la homeostasia corporal.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Este trabajo se ha realizado con el apoyo económico del Fondo de Investigación Sanitaria (PI09/91060 y PI01/00747); del CIBER de Fisiopatología de la Obesidad y Nutrición del Instituto de Salud Carlos III; de la Fundación Mutua Madrileña (AP2561/2008), y de la Fundación Endocrinología y Nutrición. Gabriel Á. Martos-Moreno recibió un contrato posformación sanitaria especializada (Río Hortega) del Instituto de Salud Carlos III (CM05/00100). Este trabajo recibió financiación del Programa Eminent Scholar del estado de Ohio, que incluye una donación de Milton & Lawrence Goll, por los proyectos del NIH DK075436-01, AG019899-06, y por la Diabetes Research Initiative y el BioMolecular Innovation and Technology Partnership de la Universidad de Ohio.

