

Sr. Editor:
El síndrome de Sturge Weber (SSW) o angiomatosis encefalotrigeminal (OMMIN#185300) es un síndrome neurocutáneo congénito, esporádico y no hereditario, con una prevalencia estimada en 1 cada 50.000 recién nacidos vivos1. Se caracteriza por un angioma plano, rojo vinoso, frecuentemente unilateral, localizado en el área de las ramas oftálmica y maxilar del trigémino (frente y párpado superior principalmente), ya presente desde el nacimiento. La lesión cutánea se acompaña homolateralmente de un angioma leptomeníngeo y en hasta un 40% de las ocasiones de un angioma coroideo. El 30-46% asocia a glaucoma, uni o bilateral.
El angioma leptomeníngeo suele afectar al lóbulo occipital, a la región parieto-occipital o incluso a todo un hemisferio. Este exceso de vascularización venosa, produce una alteración en el drenaje venoso cerebral, que conlleva estasis venosa e hipoperfusión, produciendo atrofia cerebral, y finalmente calcificaciones.
Las manifestaciones neurológicas están en directa relación con la extensión de la afectación cerebral, y son: epilepsia (casi 80%), suele iniciarse en el primer año de vida, y son habitualmente crisis parciales con tendencia a generalizarse; retardo mental (casi 60%), grave, en casi la mitad de los pacientes, y es mayor cuanto antes se produce el debut de la epilepsia, y hemiparesia contralateral a la lesión cerebral y defectos en el campo visual (hemianopsia homónima por afectación del lóbulo occipital). El diagnóstico de sospecha se basa en las calcificaciones corticales de la tomografía computarizada (TC), típicamente en líneas de ferrocarril. La confirmación diagnóstica se fundamenta en la presencia de angioma leptomeníngeo, demostrado en la resonancia magnética (RM) cerebral con contraste paramagnético, donde también se puede observar: atrofia cerebral, aumento del plexo coroideo homolateral, prominencia del diploe craneal y la malformación angiomatosa leptomeníngea que confirma el diagnóstico1, 2, 3, 4, 5. En la actualidad, se han publicado artículos donde se aplican nuevas técnicas de imagen a pacientes afectados de esta enfermedad: TC por emisión de fotón simple (SPECT), que muestra hipoperfusión en la región cortical afectada, y tomografía por emisión de positrones (PET) para ver el grado de hipometabolismo en las regiones afectadas1.
En la literatura se han descrito tres tipos de Sturge Weber: el tipo I o forma clásica, con angioma facial, leptomeníngeo y coroideo, frecuentemente con glaucoma. El tipo II, con angioma facial y sin evidencia de afectación intracraneal, y el tipo III con únicamente angiomatosis leptomeníngea1, 6, 7, 8.
Se presenta un varón de 7 años de edad, con diagnóstico por neuroimagen de SSW tipo III, tras presentar varios episodios confusionales prolongados en 72h.
Segundo hijo de padres sanos no consanguíneos, sin antecedentes familiares de migraña, ni de otras enfermedades de interés. Embarazo, parto, desarrollo psicomotor y escolarización sin incidencias.
Presenta un episodio de desconexión del medio, sin perdida de consciencia, con facies inexpresiva, desviación de la comisura bucal hacia la derecha, lenguaje incoherente y repetitivo, de una hora de duración, que cede espontáneamente, y durante el cual, no es capaz de realizar instrucciones sencillas, pero responde a alguna pregunta simple. Presenta a las pocas horas, otro episodio similar de menos de 10min de duración. El vídeo-EEG de vigilia realizado posteriormente fue normal, con ligera lentificación de la actividad de fondo.
A las 48h, presenta nuevo episodio de desconexión del medio, lenguaje repetitivo, de 20min de duración, con cefalea y sensación nauseosa. Se administran por vía intravenosa (IV) paracetamol y fenitoína, sin clara relación con el cese del episodio. No se administró diazepam, pues se pretendía comprobar si había respuesta, evitando sueño o disminución del nivel de consciencia.
A las 72h del primer episodio, presenta desconexión del medio, de una hora de duración, con lenguaje repetitivo, y respuestas incoherentes. EEG crítico durante este episodio, fue normal, con actividad de fondo enlentecida, y se vuelve a administrar fenitoína IV, sin que se pueda relacionar claramente el final de episodio, con la administración de ésta.
La exploración neurológica fuera de los episodios fue normal.
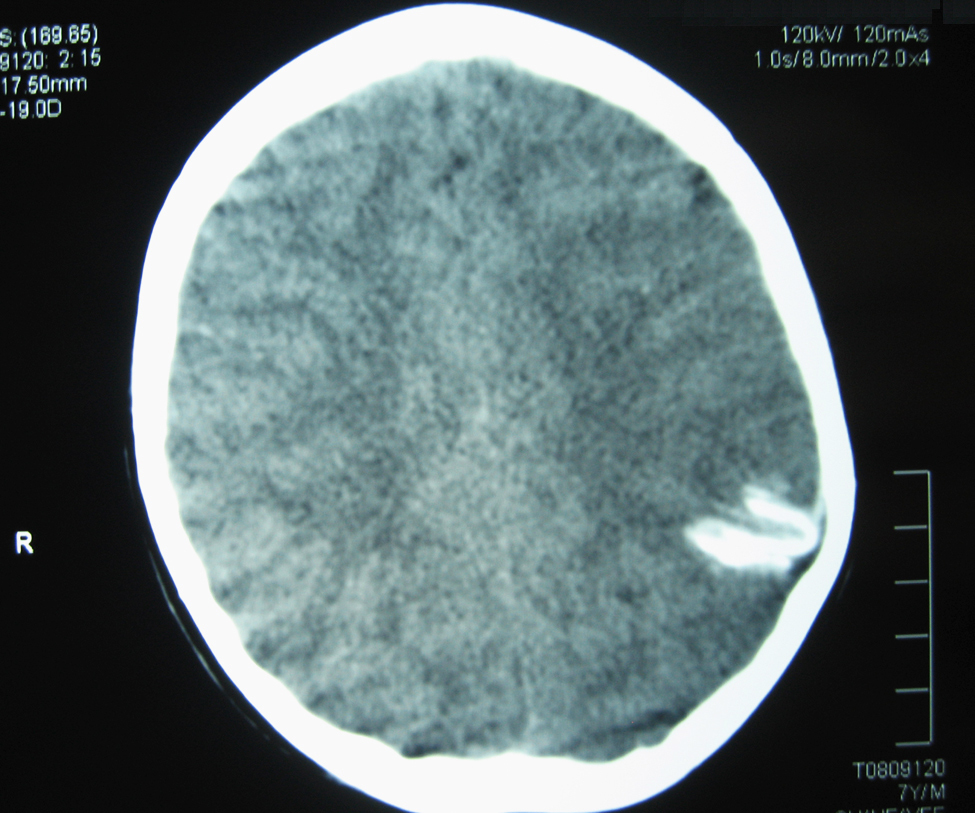
La TC craneal mostró una calcificación curvilínea con impronta en la calota craneal (Figura 1). La RM y angio-RM cerebral mostraron en área cortical parieto-occipital izquierda pequeñas áreas de hiposeñal, con leve hipotrofia y sin dilatación de arterias de mediano y gran tamaño.
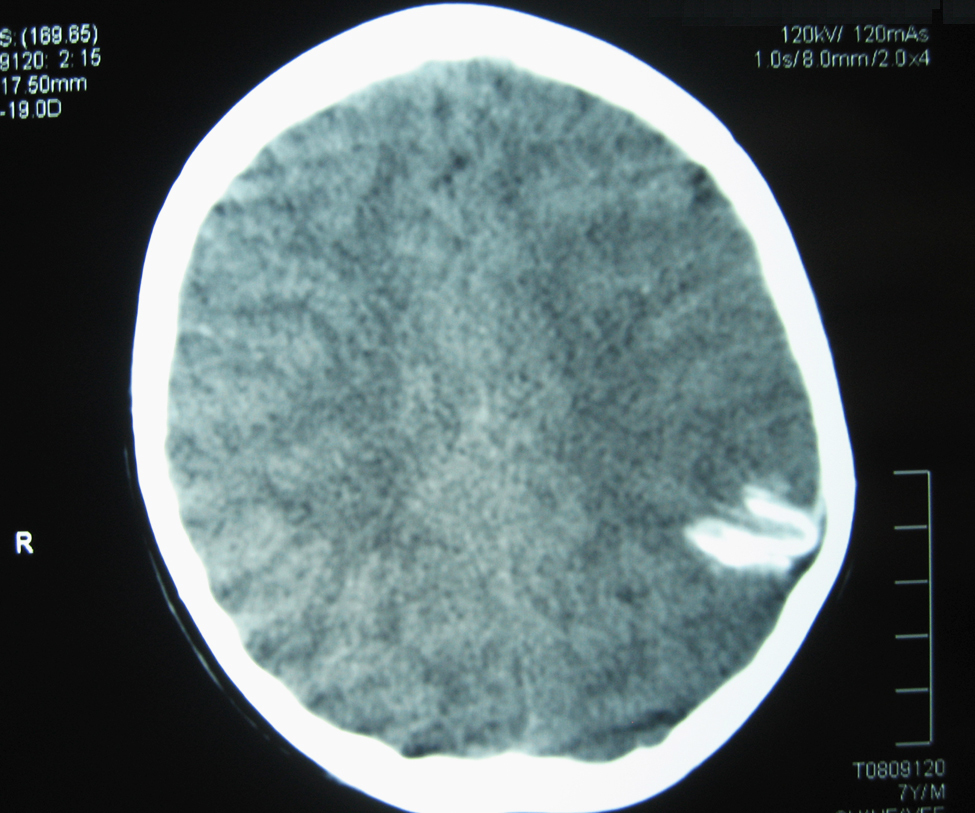
Figura 1. TC sin contraste: corte axial; se aprecia imagen hiperdensa con aspecto curvilíneo en región parieto-occipital izquierda con leve impronta sobre la calota craneal.
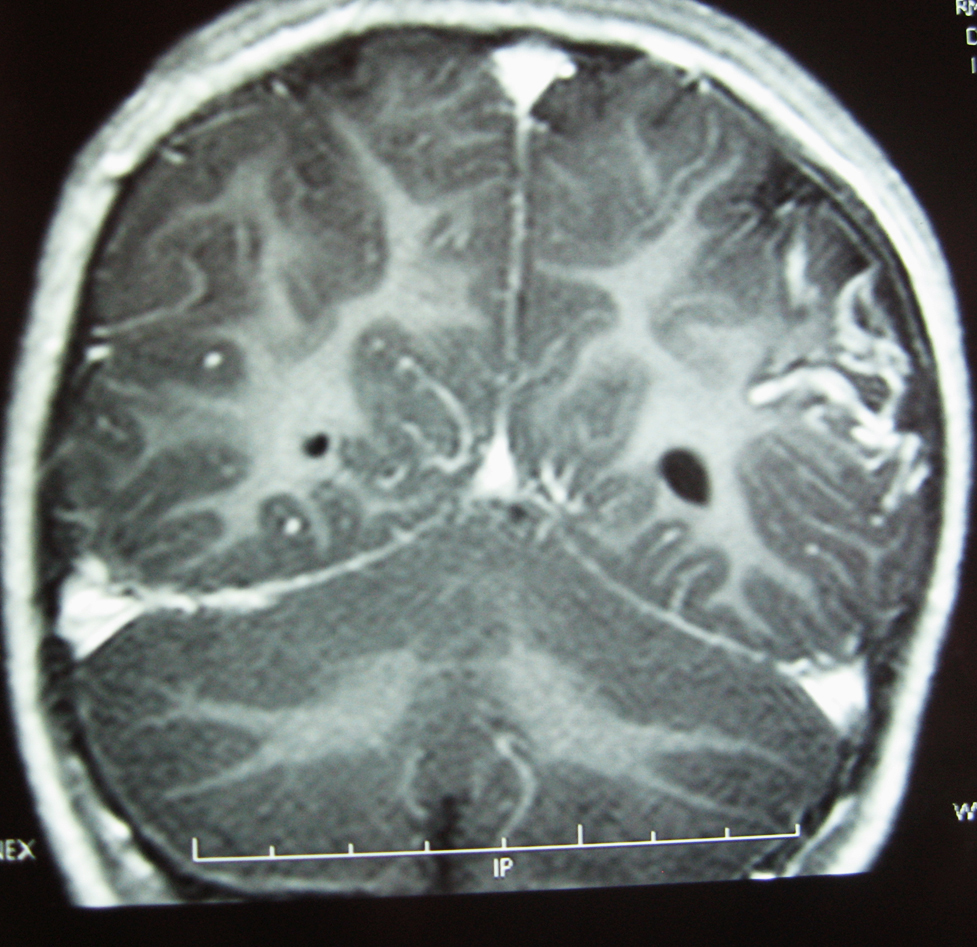
La RM cerebral con gadolinio, realizada un año después del inicio de los episodios, demostró la presencia de un angioma leptomeníngeo en la zona parieto-occipital izquierda, con atrofia cortical subyacente (Figura 2), confirmando la sospecha de SSW sin angioma facial8, 9.
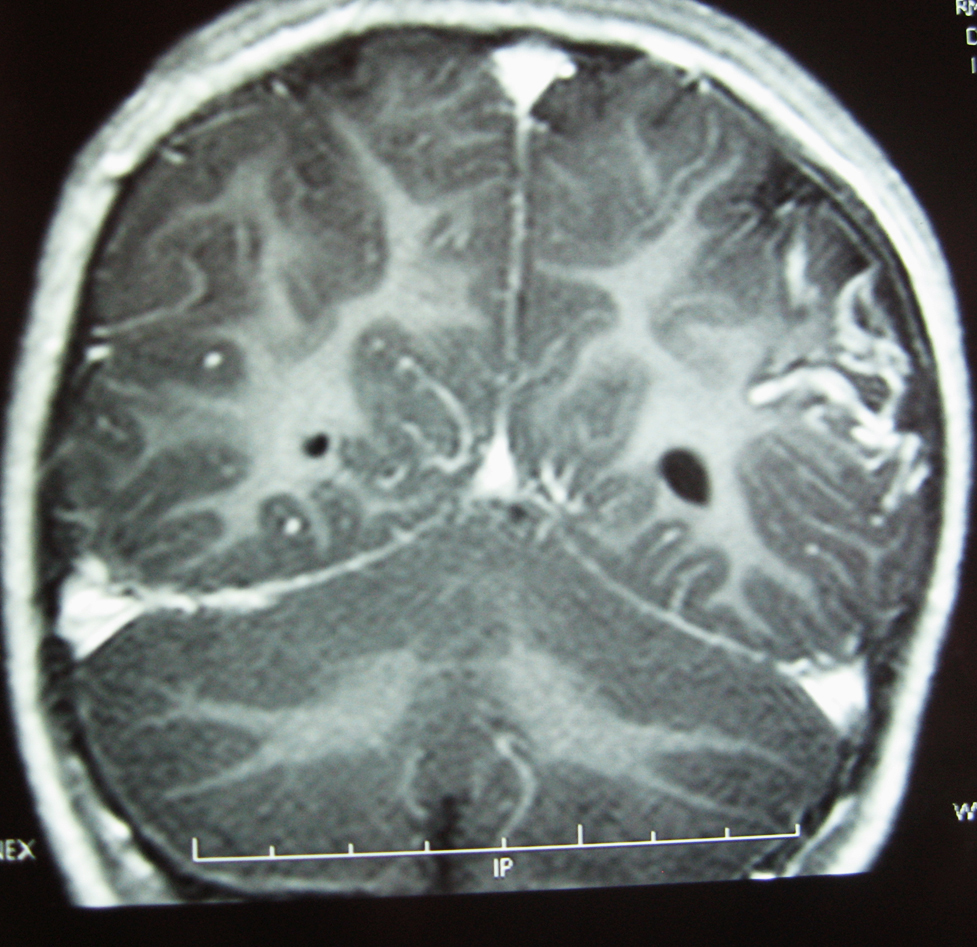
Figura 2. RM con contraste paramagnético (gadolinio) IV: corte coronal, secuencia T1; se aprecia la captación de contraste en la región parieto-occipital izquierda compatible con angioma leptomeníngeo, junto al menor tamaño hemisferio cerebral izquierdo y mayor tamaño ventrículo lateral izquierdo compatibles con hemiatrofia.
Los marcadores de enfermedad celiaca fueron negativos, y el fondo de ojo y exploración oftalmológica, normales.
Se inició tratamiento con oxcarbacepina (OXC) hasta 35mg/kg/día. Vídeo-EEG de vigilia de control a los 3 y 6 meses, normales. Permaneció libre de clínica durante 12 meses, pero posteriormente en 3 meses ha presentado 4 episodios de desconexión del medio, de menos de un 1min de duración, sin otra sintomatología. El último EEG, a los 15 meses de inicio de la clínica, mostró de forma intermitente ondas agudas y ondas lentas centroparietales bilaterales. Se aumentó la OXC a 45mg/kg/día.
Los hallazgos neurorradiológicos en nuestro paciente (calcificaciones curvilíneas parieto-occipitales con impronta en calota craneal, atrofia hemisférica y el angioma leptomeníngeo subyacente) son compatibles con SSW sin angioma facial ni coroideo5, 8, sin afectación motora ni retardo mental ni afectación de los campos visuales. La negatividad de los marcadores de enfermedad celiaca descartó el síndrome de Gobbi (epilepsia, calcificaciones intracraneales y enfermedad celiaca)10.
Las características de los episodios confusionales iniciales, con varios estatus en poco tiempo (72h), la normalidad de los EEG intercríticos y críticos, y la no respuesta a fenitoína IV en dos de los episodios, los pueden hacer compatibles con episodios confusionales causados por fenómenos isquémicos transitorios en el área cortical subyacente a la malformación angiomatosa leptomeníngea, más que a descargas epileptógenas.
En la bibliografía no hay dudas respecto a la relación entre epilepsia y el SSW, pero pocas referencias bibliográficas discuten el posible origen vascular de fenómenos neurológicos transitorios en pacientes afectados de SSW, ninguna con respecto a episodios confusionales. Sí se discuten estas posibilidades cuando los pacientes con SSW presentan migrañas con aura, como han descrito Iizuka et al11, en un paciente adulto afecto de SSW que presentaba migrañas con auras prolongadas de posible origen isquémico. Jansen et al12 presentan 2 pacientes con parálisis unilaterales transitorias y SSW, uno de ellos debida a descargas epileptógenas, con EEG crítico alterado, y el otro debido a supuestos fenómenos isquémicos, enfatizando lo difícil que resulta al clínico distinguir el origen isquémico o epiléptico de este tipo de episodios, así como la importancia del EEG crítico. Dora et al13 presentan un paciente con SSW y migraña hemipléjica transitoria debida también a fenómenos vasculares. Kossoff et al14 en su revisión describen que el inicio de la epilepsia en la infancia en pacientes afectados de SSW puede ser, en ocasiones (22-50%), agrupada en un patrón de varias crisis en poco tiempo o con crisis prolongadas, alternando con periodos prolongados libres de clínica, sin clara relación con la medicación, y que funcionalmente podría estar relacionado con periodos de isquemia focal transitoria.
Inicialmente, antes del diagnóstico de SSW, se planteó el diagnóstico diferencial entre migraña confusional y epilepsia, por lo que, dada la duración de los episodios, se inició tratamiento con OXC para cubrir la posibilidad de crisis parciales y se mantuvo dada la desaparición de los episodios y las dudas respecto a su origen. Creemos interesante destacar que los pacientes afectados de SSW pueden presentar trastornos paroxísticos de tipo isquémico y no epiléptico, lo que puede ser de difícil diferenciación. Pensamos que los episodios iniciales pudieron ser de origen isquémico secundario a robo vascular, pero que posteriormente, el paciente ha desarrollado una epilepsia sintomática, dadas la afectación cortical, que aparece más atrófica en la neuroimagen de control al año del inicio de los síntomas, las alteraciones del EEG, que aparecen también a partir del año, y las características de los nuevos episodios críticos, desconexión del medio de escasos segundos de duración. No se instauró tratamiento antiagregante en su momento, al considerar que el fenómeno isquémico fue secundario a fenómeno de robo vascular, pero en función de la evolución nos lo replantearemos, dado que está descrito que la estasis venosa en la malformación angiomatosa puede favorecer la trombosis12, 15, 16.
Autor para correspondencia. jlopezpi@salud.aragon.es

