

La deficiencia de succínico semialdehído deshidrogenasa o aciduria gammahidroxibutírica (GHB) es una rara enfermedad autosómica recesiva por alteración en el metabolismo del neurotransmisor inhibidor del ácido gammaaminobutírico. Debido a la deficiencia de esta enzima, se produce acúmulo del metabolito ácido GHB. El espectro clínico es heterogéneo con distintas manifestaciones neurológicas. El tratamiento más utilizado es la vigabatrina, aunque se discute su eficacia. Presentamos el caso de una familia con 2 hijos afectados. El mayor presenta retraso en la adquisición de hitos motores, marcha liberada a los 2 años, dificultad en la psicomotricidad fina, hipotonía axial, normorreflexia y retraso en el lenguaje tanto comprensivo como expresivo. Se evalúa al menor por hipotonía axial. Estudio metabólico: aumento de ácido GHB en orina y en plasma. El diagnóstico se confirma mediante el análisis de mutaciones del gen ALDH5A1. Se trata con vigabatrina en dosis bajas, lo que conduce a una disminución de los niveles de GHB en plasma en más de dos tercios y una evolución clínica favorable.
Succinic semialdehyde dehydrogenase deficiency (gamma-hydroxybutyric aciduria) is a rare neurometabolic disease caused by a deficiency in gamma-aminobutyric degradation, resulting in an increase in gamma-hydroxybutyric acid in biological fluids. The clinical spectrum is heterogeneous, including a variety of neurological manifestations and psychiatric symptoms. The treatment usually used is vigabatrin, but its clinical efficacy is under discussion. We present two affected siblings. The older brother was examined when he was 2.5 years old due to psychomotor and developmental delay, disturbances in motor coordination, axial hypotonia and language disability. His younger brother had mild axial hypotonia when 5 months old. Metabolic studies demonstrated a high plasma and urine concentration of gamma-hydroxybutyric acid. Mutation analysis of the gene ALDH5A1 confirmed the disease. After 1 year of treatment with low-doses of vigabatrin of the older patient, a decrease in gamma-hydroxybutyric acid plasma levels and a slow clinical improvement were observed.
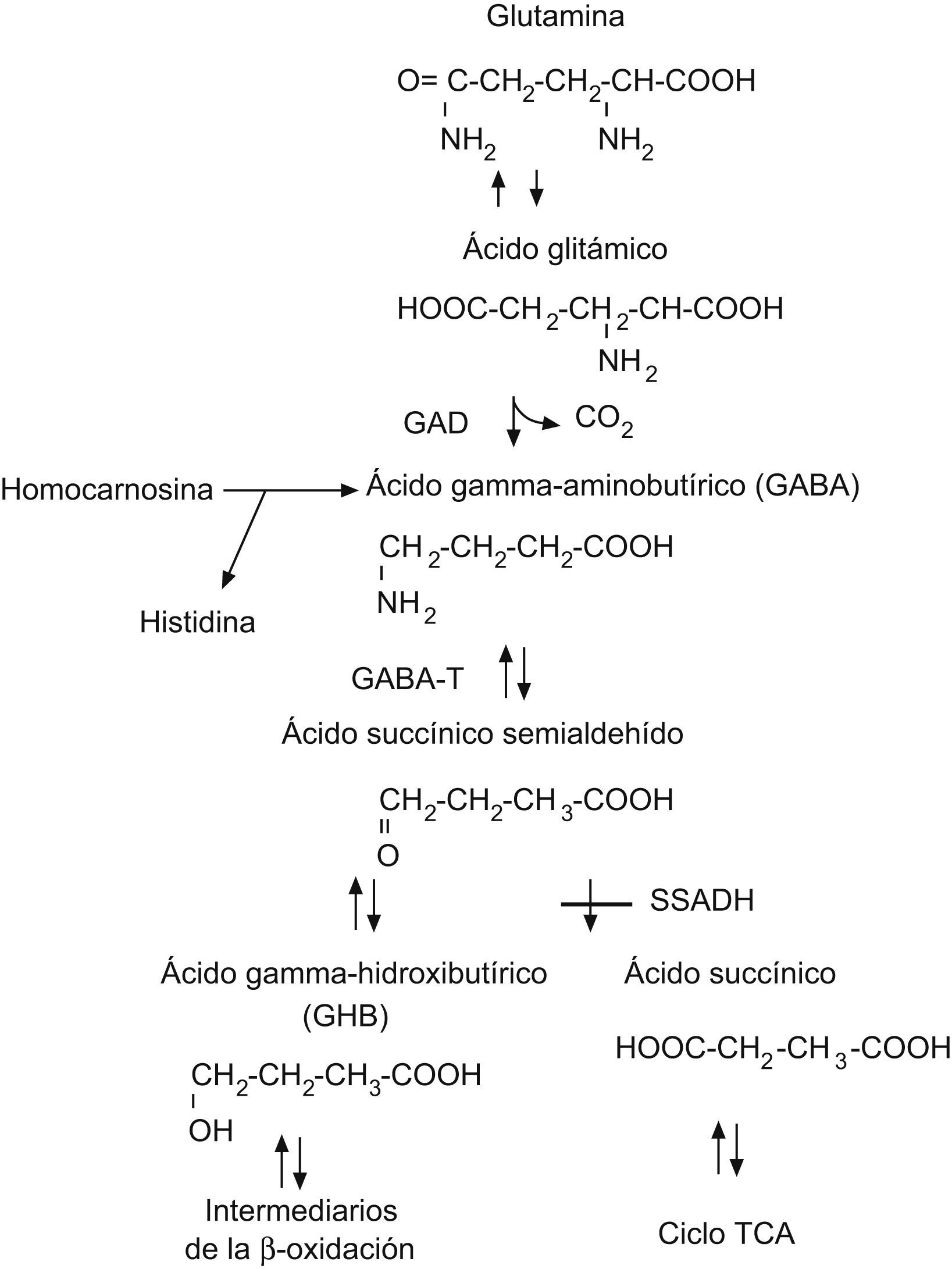
La deficiencia de succínico semialdehído deshidrogenasa (SSADH) es una enfermedad rara, autosómica recesiva, de la vía de degradación del neurotransmisor inhibidor del ácido gammaaminobutírico (GABA)1 mediante una transaminación, el GABA se convierte en ácido succínico semialdehído que, a su vez, se degrada a ácido succínico por acción de la enzima SSADH. Cuando se produce una deficiencia genética de esta enzima, el ácido succínico semialdehído se reduce a ácido gammahidroxibutírico (GHB), agente neurotóxico que se acumula en fluidos fisiológicos (fig. 1). La elevación de GHB puede medirse tanto en orina como en sangre y líquido cefalorraquídeo, y es la clave para el diagnóstico de esta enfermedad2; sin embargo, aunque la alteración metabólica está bien documentada, no se conoce con suficiente claridad cómo el exceso de GHB contribuye a los diversos fenotipos clínicos neurológicos. El gen ALDH5A1, cuyo defecto es causante de la enfermedad, se ha identificado en el brazo corto del cromosoma 63; hay numerosas mutaciones y existe una pobre correlación genotipo-fenotipo4.

El espectro clínico de la deficiencia de SSADH incluye manifestaciones como retraso mental, hipotonía, hiporreflexia, ataxia no progresiva, autismo, alteraciones del desarrollo del lenguaje (dispraxia, alteración tanto del lenguaje comprensivo como expresivo, etc.) y alucinaciones5–7. Hasta en la mitad de los casos se asocian crisis epilépticas8. Los estudios de EEG pueden ser normales en hasta la mitad de los casos. Estudios de neuroimagen, como la resonancia magnética nuclear (RMN) cerebral, muestran normalidad en hasta un tercio de los casos, y ésta puede presentar hiperintensidad en T2 en los globos pálidos, la sustancia blanca y los núcleos dentados y atrofia cerebelosa9,10. No hay tratamiento establecido para la deficiencia de SSADH y existe controversia en la utilización de vigabatrina, ya que su eficacia clínica a largo plazo es discutible11,12. Presentamos una familia con 2 hijos afectados de deficiencia de SSADH, su diagnóstico, tratamiento y evolución clínica.
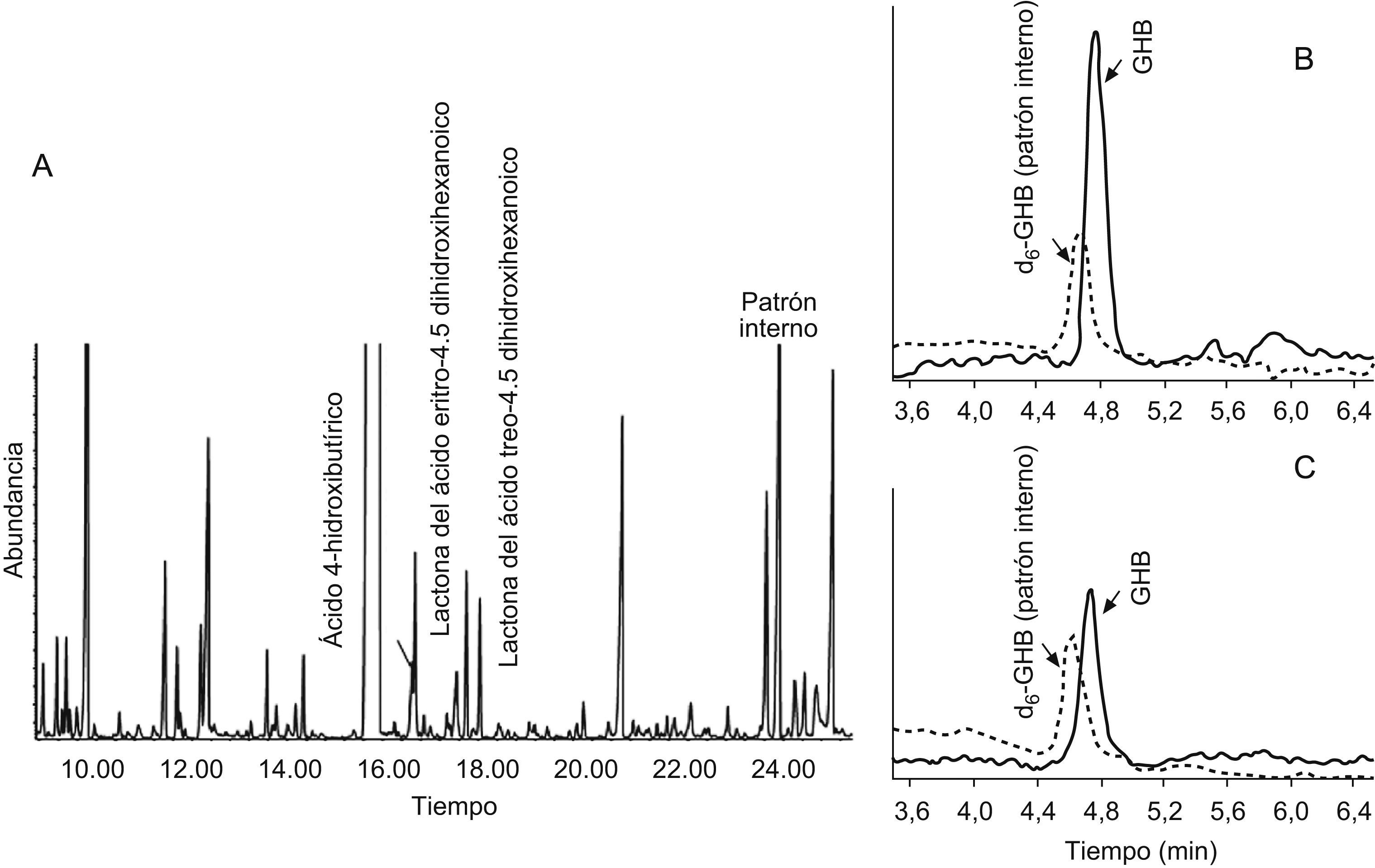
Caso clínicoMadre con cierto retraso psicomotor, deambulación liberada a los 20 meses, retraso en el lenguaje expresivo y dificultades para el aprendizaje. Padre sano. Sin consanguinidad. Se examina al hijo mayor por retraso psicomotor. El embarazo había sido normal y el parto por cesárea por sospecha de pérdida de bienestar fetal. Sin reanimación. Había presentado retraso en la adquisición de hitos motores, marcha liberada a los 2 años, torpeza motriz, dificultad en la psicomotricidad fina, hipotonía axial y normorreflexia; trastorno del lenguaje con retraso tanto a nivel comprensivo como expresivo. A los 5 años sigue órdenes que implican 2 acciones, amplía su vocabulario e intenta construir frases sencillas y le cuesta mantener la atención. No ha presentado crisis epilépticas. La analítica en sangre y orina, que incluye hemograma, hitachi, perfil hepático, CK y TSH, es normal. El estudio metabólico (aminoácidos, test de saicar para el déficit de adenilosuccinato liasa, porcentaje de transferrina deficiente en hidratos de carbono para los defectos congénitos de glucosilación y actividad biotinidasa) es normal, salvo el análisis de ácidos orgánicos en orina, donde se observa una excreción muy aumentada de GHB, compatible con aciduria GHB por deficiencia de la enzima SSADH (fig. 2A). Los niveles de GHB en plasma, cuantificados por HPLC/MS/MS con dilución de isótopos estables también son muy elevados: 145μmol/l (VC<10) (fig. 2B). Mediante el análisis de mutaciones del gen ALDH5A1 en ADN, se identifican 2 mutaciones patogénicas ya descritas en distintos alelos: c.278G>T y c.621delC.
 Análisis de ácidos orgánicos en orina por cromatografía de gases y espectrometría de masas. B) Análisis de gammahidroxibutírico (GHB) por HPLC/MS/MS en plasma al diagnóstico. C) Análisis de GHB por HPLC/MS/MS en plasma al año de tratamiento con vigabatrina.")
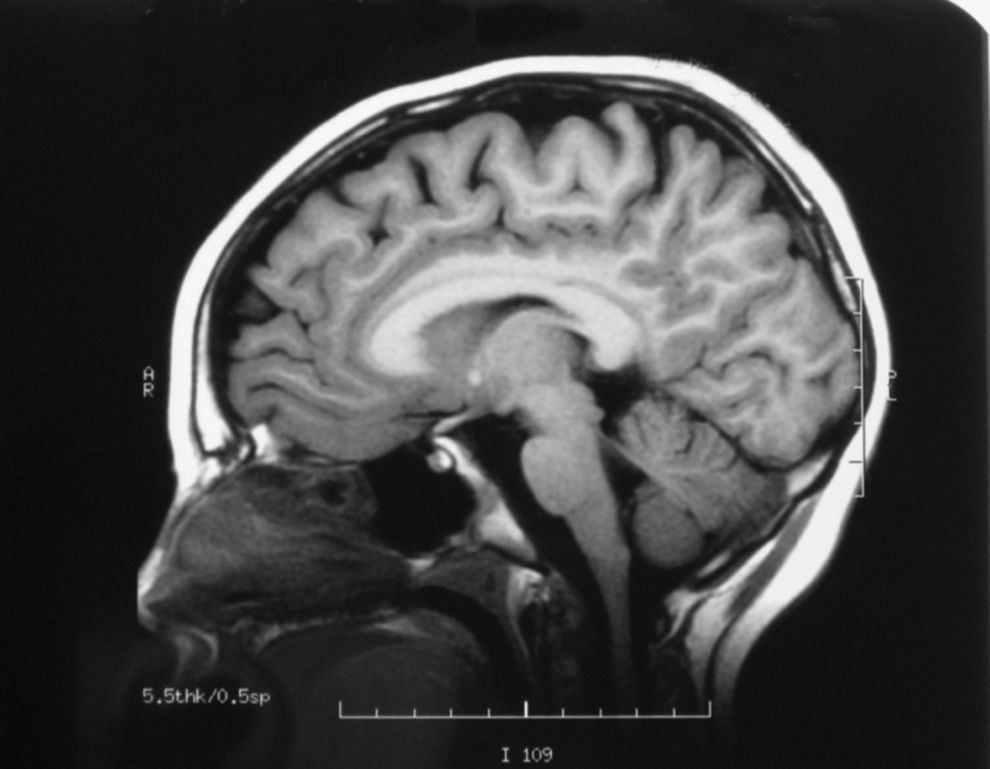
El EEG es normal y la RMN cerebral muestra una leve atrofia difusa (fig. 3), mientras que la espectroscopia muestra normalidad metabólica de la sustancia gris cortical y afectación de la sustancia blanca subcortical.

Previo consentimiento, se inicia tratamiento con dosis bajas de vigabatrina (20mg/kg/día). Al año, se observa una bajada cuantitativa en sangre de GHB hasta 55μmol/l (fig. 2C) y una evolución clínica lenta, aunque favorable; persiste hipotonía, alteración de la psicomotricidad fina y del desarrollo del lenguaje. No presenta dificultades visuales ni retinopatía. Se decide suspender el tratamiento a petición familiar.
El hermano pequeño acude a la consulta por hipotonía axial. Se confirma un aumento en orina de GHB, pero el resto del estudio analítico es normal. Presenta una evolución más favorable que el hermano. A los 19 meses consigue marcha con ayuda de una mano y consigue incorporarse desde el suelo con apoyo. Se aprecian dificultades psicomotoras tanto a nivel grosero como fino, la hipotonía persiste con normorreflexia. Se encuentran dificultades en el lenguaje con retraso sobre todo a nivel expresivo, no emite palabras, emite algún bisílabo referencial, aunque impresiona su buena comprensión. En ningún momento ha presentado crisis comiciales.
DiscusiónLa deficiencia de SSADH es un error metabólico hereditario causado por una alteración en la vía de degradación del GABA13. Debido a la deficiencia enzimática, los pacientes acumulan GHB en fluidos biológicos, pero no presentan acidosis metabólica ni cetonuria, hiperamonemia o hipoglicemia (típicas de otras acidurias orgánicas). En nuestros pacientes, el diagnóstico de laboratorio se llevó a cabo mediante el análisis de ácidos orgánicos en orina por cromatografía de gases-espectrometría de masas14. El GHB es un metabolito volátil y su detección es complicada, por lo que esta enfermedad podría estar infradiagnosticada. Sin embargo, la presencia en fluidos fisiológicos de los isómeros treo- y eritro- del ácido 4,5-dihidroxihexanoico, que se producen por la condensación del ácido succínico semialdehído con el ácido pirúvico, es patognomónica y característica de esta enfermedad15. El análisis con isótopos estables del GHB en fluidos fisiológicos es el método más preciso para la cuantificación y la monitorización de sus niveles durante el tratamiento16,17. En uno de nuestros pacientes se pudo demostrar una disminución de los niveles de GHB en sangre al año de tratamiento con vigabatrina, como ya se había descrito en otros casos18. La identificación de 2 mutaciones patogénicas del gen ALDH5A 1 en diferentes alelos (estudio familiar) del ADN del paciente confirmó la enfermedad. El cambio c.278G>T en el exón 1 da lugar al cambio aminoacídico de una cistina por una fenilalanina en la posición 93 (c.Cys93Phe). El análisis de expresión de esta mutación en las células de mamífero HEK 203 demostró una actividad del SSADH muy disminuida (el 3% del alelo salvaje). La otra mutación identificada es c.621delC en el exón 4 y da lugar a un cambio del aminoácido serina por valina en la posición 208 y a un codon de parada de la proteína (p.Ser208ValfsX3), lo que presumiblemente produce una proteína truncada4. Debido al bajo número de pacientes con genotipos idénticos, no se ha podido establecer una relación fenotipo-genotipo en esta enfermedad. Aunque en principio, por la gravedad del combinado genético de nuestros pacientes cabría esperar un fenotipo grave, existen factores adicionales metabólicos y genéticos que pueden influir en las manifestaciones clínicas de la enfermedad.
El espectro clínico de la deficiencia de SSADH es heterogéneo, incluye retraso mental, hipotonía, hiporreflexia, ataxia no progresiva, autismo, alteraciones del desarrollo del lenguaje (dispraxia, alteración tanto del lenguaje comprensivo como expresivo, etc.), síntomas neuropsiquiátricos, alteraciones del sueño, hiperactividad, inatención, ansiedad y alucinaciones5,6. Se han descrito pacientes con manifestaciones extrapiramidales que incluyen corea, mioclonía y distonía. Nuestros pacientes presentan, hasta la fecha, una clínica relativamente leve con retraso psicomotor y alteración del lenguaje, sin crisis epilépticas. El estudio de neuroimagen (RMN cerebral) del hermano mayor realizado a los 8 años de edad mostró atrofia, como en otros casos ya descritos. El estudio de espectroscopia en el paciente reveló normalidad metabólica de la sustancia gris cortical y afectación de la sustancia blanca subcortical en términos de metabolismo energético deficitario y empobrecimiento glial sin signos de acumulación de lactato, datos inespecíficos no relacionados con el déficit metabólico, como se describe en la literatura médica.
El tratamiento de la deficiencia de SSADH es controvertido y hasta la fecha no se ha encontrado un tratamiento plenamente satisfactorio, aunque en principio, el uso de vigabatrina parece la opción más evidente, ya que este compuesto es un inhibidor de la GABA aminotransferasa e impide el paso a ácido succínico semialdehído, precursor del GHB19. Está publicado que la administración de vigabatrina en bajas dosis (20–25mg/kg/día) conduce a la disminución de los niveles de GHB con cierta mejoría clínica a nivel cognitivo, y que dosis más elevadas del fármaco (75mg/kg/día) producen empeoramiento en el registro EEG y crisis epilépticas así como alteración irreversible en el campo visual20. Tratamos a nuestro paciente durante un tiempo en dosis bajas y presentó una fuerte disminución de los niveles de GHB en sangre con una lenta mejoría clínica, aunque persistió una alteración del desarrollo del lenguaje. Debido a los posibles efectos secundarios descritos con la vigabatrina, se suspendió el tratamiento. La utilización de otros fármacos, como la taurina, que se ha utilizado en un paciente de 2 años y medio con resultado de mejoría en el comportamiento, interacción social, marcha y coordinación21 y el antagonista del receptor de GABA (B) SGS-742, que se ha probado en ratones con deficiencia de SSADH con buenos resultados22, necesita más estudios y ensayos clínicos para poder llegar a conclusiones definitivas. Otra pauta terapéutica ha sido el uso de lamotrigina, cuyo efecto farmacológico bloquea los aminoácidos excitatorios, especialmente el mayor precursor GABA-érgico: el ácido glutámico. Se usó satisfactoriamente en un paciente con deficiencia de SSADH, en el que la vigabatrina indujo crisis epilépticas. También se ha indicado la eficacia de la dieta cetogénica para el control de las crisis epilépticas y el retraso del comienzo de la ataxia23. En cualquier caso, en la actualidad, el efecto tan limitado y controvertido del tratamiento obliga a hacer hincapié en la importancia del tratamiento psicopedagógico y del lenguaje24.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

