

El retraso generalizado del desarrollo o retraso madurativo hace referencia a aquellos niños menores de 5 años con discapacidad intelectual en los que, dada la dificultad para realizar pruebas psicométricas, el diagnóstico de retraso mental no puede ser preciso1. Identificar la etiología de este retraso continúa siendo un reto en la actualidad a pesar de los avances en genética, neuroimagen y enfermedades metabólicas.
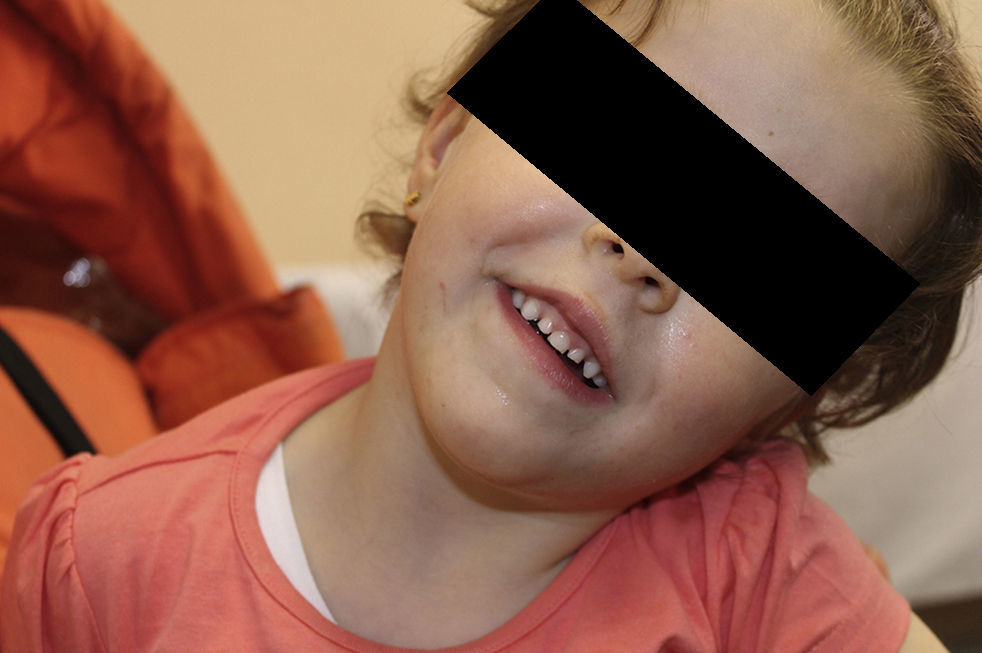
Presentamos el caso clínico de una niña de 3 años y 8 meses, en seguimiento en consultas de Neurología por retraso generalizado del desarrollo. Remitida a los 10 meses de edad desde Oftalmología con los diagnósticos añadidos de hipoplasia del nervio óptico derecho y estrabismo convergente bilateral. Entre los antecedentes familiares, obstétricos y perinatales no hay ningún dato de interés. Presenta un retraso leve en los hitos del desarrollo psicomotor desde las primeras etapas. Durante el seguimiento logra la marcha libre a los 18 meses, mantiene un retraso manipulativo y del lenguaje, siendo más marcado a nivel expresivo. Destaca un fenotipo peculiar con frente prominente, fisuras palpebrales descendentes, narinas antevertidas y paladar ojival (fig. 1); siendo el resto de la exploración física y somatometría normales. Se objetiva tendencia a mantener la boca entreabierta y la aparición de un incisivo único superior, confirmándose, tras realización de Rx facial, la presencia del incisivo definitivo.

Se realizan analítica sanguínea con perfil metabólico y hormonas tiroideas, RM craneal con espectroscopia, EEG, potenciales evocados auditivos y visuales del tronco cerebral; cariotipo y estudio de reordenamientos subteloméricos y de trastornos genómicos recurrentes por MLPA, todos ellos con resultado de normalidad.
Posteriormente, se amplía el estudio para descartar anomalías submicroscópicas mediante la técnica de array CGH, empleándose un array genómico personalizado enriquecido en más de 380 regiones relacionadas con síndromes de microdeleción/microduplicación, regiones subteloméricas y pericentroméricas y genes relacionados con discapacidad intelectual y alteraciones del desarrollo (KaryoArray®, basado en la plataforma Agilent de 8×60k)2. Esta técnica permitió identificar una alteración genómica de aproximadamente 379,48Kb en el cromosoma 3, región 3p13, que provoca la pérdida de una de las 2 copias del gen FOXP1.
Las proteínas con dominios FOX parecen actuar como factores de transcripción, modulando la activación de otros genes mediante su unión con el ADN3. El gen FOXP1 aparece citado por primera vez en un artículo de 2001, junto a FOXP2, donde identifican estos 2 nuevos genes actuando como represores transcripcionales en el pulmón de ratones3. En octubre de ese año aparece la primera relación entre genes FOXP, concretamente FOXP2 y el habla4. La unión al ADN de esta proteína FOXP2 está condicionada por su interacción con otras moléculas de FOXP2, pero también a la formación de heterodímeros con FOXP1 o FOXP45, siendo posible que la mutación de estos genes diese lugar también a alteraciones del lenguaje y del habla.
Han sido publicados en la literatura varios pacientes con pérdida de una de las 2 copias del gen FOXP1 de novo6–10. Estos pacientes presentaban retraso mental o generalizado del desarrollo, con antecedentes perinatales sin incidencias. Todos tuvieron retraso en el desarrollo motor grueso; empezaron a hablar tardíamente, estando el lenguaje expresivo más afectado que el comprensivo. Algunos tuvieron tendencia a mantener la boca entreabierta y dificultades para la deglución; siendo frecuentes también, problemas comportamentales como hiperactividad y rasgos del espectro autista. Varios de estos pacientes presentaban una frente prominente, implantación del pelo frontal hacia arriba y fisuras palpebrales descendentes. La RM y el EEG resultaron normales en la mayoría de los pacientes descritos, encontrándose un caso con malformación Chiari tipo I, hipoplasia cerebelosa, cuerpo calloso dismórfico y descargas epilépticas en el EEG9, y otro, con ventrículos cerebrales prominentes10. Así, la deleción o pérdida de una de las 2 copias de FOXP1 no solo se relacionaría con alteraciones verbales, sino que conduciría a un desarrollo anormal de estructuras cerebrales implicadas en procesos cognitivos y psicomotrices8. La deleción aislada de FOXP1 parece relacionarse por tanto con retraso mental, afectación motora y del lenguaje, principalmente expresivo y, con un fenotipo peculiar como el descrito en nuestra paciente.
El examen dismorfológico continúa teniendo un alto rendimiento diagnóstico pudiendo orientarnos a genes o mutaciones específicas, sobre todo, dada la falta de disponibilidad y el alto coste de algunas pruebas genéticas. De esta forma, parece interesante descubrir el fenotipo clínico de enfermedades descritas recientemente gracias a la genética. Así, al examen físico y exploración neurológica, hemos de añadir este examen dismorfológico, porque en muchos casos anomalías menores pueden evidenciar un diagnóstico etiológico1.

