

La mucolipidosis tipo II o enfermedad de células de inclusión (OMIM 252500) es una rara enfermedad de deposito lisosomal con carácter de herencia autosómico recesivo, causada por la deficiencia de la enzima uridin difosfato (UDP) N-acetilglucosamina: N-acetilglucosamina-1fosfotransferasa (GlcNAc-fosfotransferasa-EC2.7.8.17)1,2; esta enzima cataliza el paso inicial en el marcaje de la manosa 6 fosfato (M6P) en la síntesis de nuevas enzimas lisosomales, donde se transfiere un grupo GlcNAc-1-fosfato a la manosa seleccionada sobre el oligosacárido de la nueva hidrolasa sintetizada2–4. Las enzimas lisosomales, modificadas con M6P, se unen al receptor de M6P en trans-Golgi y son translocadas al endosoma y posteriormente al lisosoma2. Cuando GlcNAc-fosfotransferasa es deficiente se altera la síntesis y trasporte de las hidrolasas hasta el lisosoma, por lo cual se acumula material no degradado de lípidos y mucopolisacáridos, formando los depósitos lisosomales, reconocidos claramente como la causa de las manifestaciones propias de la entidad1.
La incidencia varía según la población evaluada. Se estima que en Portugal es de 1/120.000 recién nacidos vivos5, de 1/252.000 recién nacidos vivos en Japón6 y de 1/642.000 recién nacidos vivos en Holanda7, siendo frecuente en otras poblaciones como la canadiense (región noreste de Quebec), donde se estima una frecuencia de 1/6.184 recién nacidos vivos y una frecuencia de portadores de 1/398.
Dentro de las principales manifestaciones clínicas se encuentra retraso psicomotor severo, baja talla, facies tosca, hiperplasia gingival, corneas claras9, hepatoesplenomegalia, disostosis múltiple, contracturas articulares1,10, infecciones respiratorias a repetición, hipotonía generalizada, piel gruesa y rígida, cardiomegalia5, hernias inguinales y umbilical9. El diagnóstico se confirma bioquímicamente mediante la medición de la actividad enzimática de las hidrolasas lisosomales, que se encuentra normal o elevada en fluidos extracelulares como suero y disminuida en cultivo de fibroblastos o por la medición directa de la actividad de la UDP-N-acetilglucosamina 1-fosfotransferasa10. La deficiencia total o casi total de GlcNAc-fosfotransferasa resulta en la ausencia de marcaje lisosomal en muchos tipos de células y tejidos2.
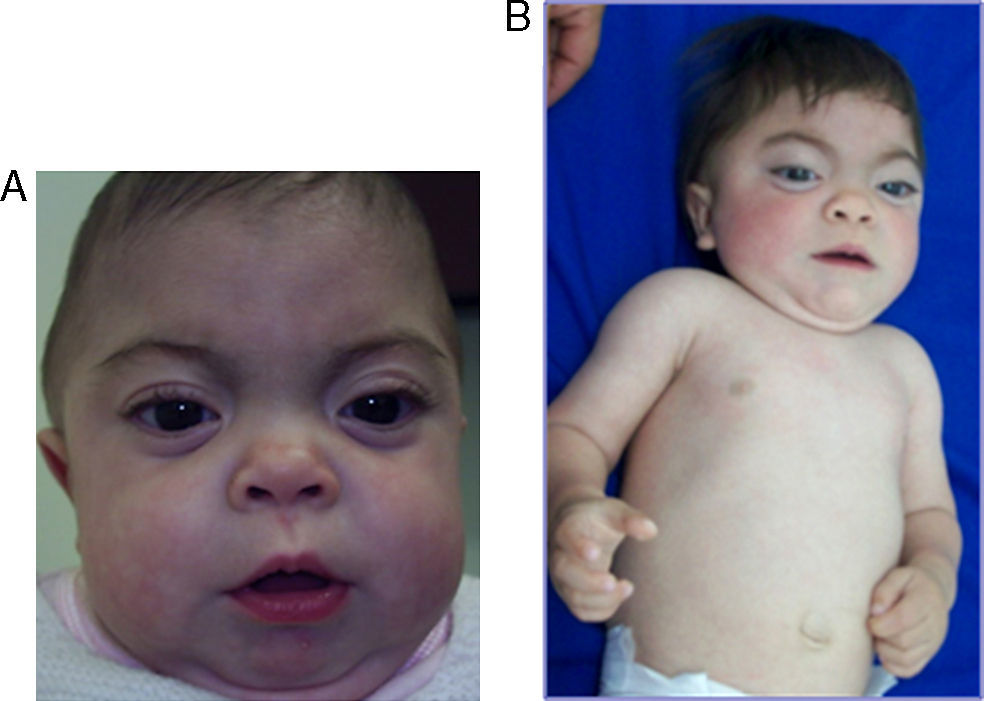
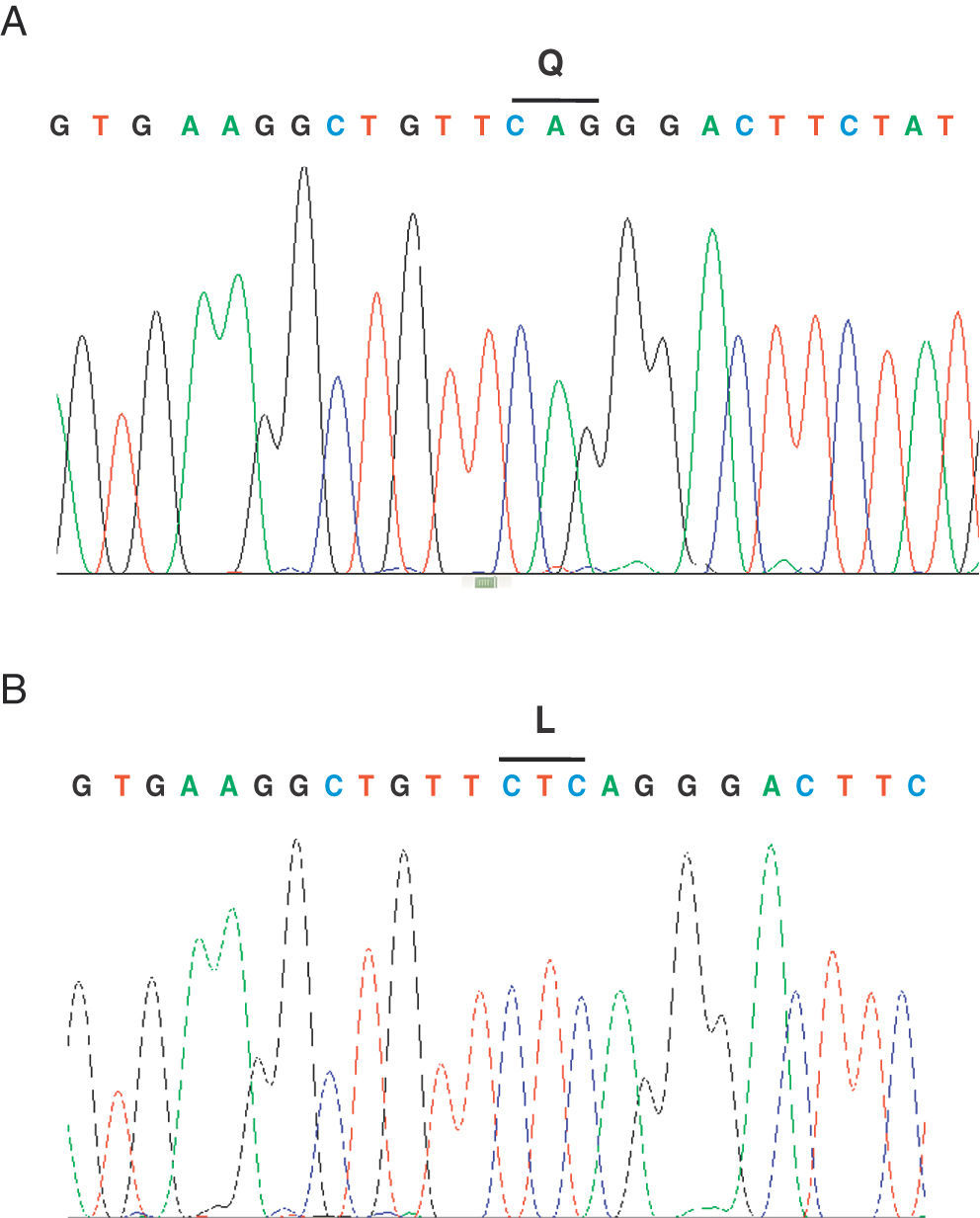
Se describe el caso de un paciente de sexo femenino, de 27 meses de edad, sin historia de consanguinidad parental, natural de Onzaga-Santander-Colombia, consulta a los 5 meses de edad por cuadro de retraso de desarrollo psicomotor, facies tosca, infecciones respiratorias a repetición y antecedente de hermano mayor con cuadro clínico similar que fallece a la edad de 2 meses sin diagnóstico. Al examen físico se evidencia fenotipo hurleriano dado por baja talla, facies tosca, hipertrofia gingival, párpados esponjosos, córneas claras, cuello corto, marcada limitación articular, mano en garra, displasia de cadera, hernia umbilical, hepatomegalia leve y cifoescoliosis lumbar (fig. 1 A y B). La paciente presenta excreción aumentada de sulfato de queratan y condroitín en el análisis de mucopolisacariduria. La actividad de varias enzimas lisosomales se encuentra elevada en suero: α-L-iduronidasa 4,5 veces el valor normal, arilsulfatasa A 3 veces, iduronato 2-sulfatasa 5,5 veces, β-glucuronidasa 4 veces, hesoxaminidasa A 2,2 veces y α-manosidasa 4 veces. Se realiza el estudio molecular por medio de secuenciación directa de los exones del gen GNPTAB; se identifica una mutación en el exón 19; c.3503_3504delTC, la cual causa una mutación tipo Frameshift que produce un codón prematuro de parada (p.L1168QfsX5) (fig. 2) originando una proteína truncada. Adicionalmente, se realizó el estudio molecular a los demás miembros de la familia, por medio de la técnica de RFLP, confirmándose el estado portador en los padres y hermano mayor.
 y 27 meses (B): facies toscas, párpados esponjosos, córneas claras, limitación a la extensión articular y hernia umbilical.")
 y de un control sano (B). Se muestra la mutación 3503_3504delTC del exón 19 de la paciente en estudio.")
La mutación identificada causa un codón de parada prematuro, resultando en deleción de 81 aminoácidos en la subunidad β, hacia la región C-terminal de la proteína, llevando a la pérdida del segundo dominio trasmembrana7, lo cual altera severamente el producto proteico y origina la enfermedad2,4,10,11. Kudo et al.2 demostraron con el estudio funcional de la mutación homocigota 3503_3504 del TC que no hay producción de actividad de GlcNAc-fosfotransferasa. Esta mutación ha sido descrita como la más frecuente en los pacientes con mucolipidosis tipo II en población canadiense y la mutación más frecuente en portadores de la región noreste de Quebec (frecuencia de portadores 1/39 individuos)1,8.
Comunicamos así el primer caso de mucolipidosis tipo II confirmado molecularmente en población colombiana; la paciente presenta fenotipo hurleriano descrito clásicamente en la enfermedad. Llama la atención el patrón cromatográfico de excreción de mucopolisacaridos en orina característico de síndrome de Morquio (sulfato de queratan), con franca elevación de enzimas lisosomales en sangre. Esta discordancia en la clínica y los estudios bioquímicos deben hacer pensar en diagnósticos diferenciales de las mucopolisacaridosis, como las mucolipidosis. En este caso específico, mucolipidosis tipo II. En cuanto a la actividad enzimática de GlcNAc-fosfotransferasa en fibroblastos, en este caso no se realizó debido a las dificultades técnicas de este.

