

Clásicamente los síndromes Say-Barber-Biesecker-Young-Simpson (SBBYSS) y genitopatelar (SGP) son entidades clínicas distintas causadas por variantes de truncamiento de novo del gen KAT6B. De penetrancia completa y expresividad variable, existen rasgos fenotípicos comunes y otros característicos (tabla 1). Actualmente, la literatura mundial recoge 89 casos confirmados: 58 SBBYSS, 18 SGP y 13 de fenotipo intermedio1.
Características clínicas
| SGP | SBBYSS | Ambos | |
|---|---|---|---|
| Esqueléticos | Contracturas de flexión en las caderas y las rodillasPie zamboAnomalías en costillas, pelvis y columna vertebral | Pulgar y primer dedo del pie alargados | Agenesia o hipoplasia de la rótula |
| Neurológicos | Agenesia del cuerpo calloso | Déficit intelectualHipotoníaPérdida de audición bilateral | |
| Genitourinarios | Quistes renalesHidronefrosisClitoromegalia o hipoplasia de los labios mayores y/o menores en mujeresHipoplasia escrotal | Criptorquidia | |
| Gastrointestinales | Atresia o estenosis analDuplicación rectalPosición anterior del ano | Hendidura palatina | Malrotación del intestino delgadoReflujo gastroesofágico |
| Cardiacos | Cardiopatías congénitas: septo auricular, septo ventricular, ductus arterioso persistente o foramen oval persistente | ||
| Dismórficos | Microcefalia | BlefarofimosisPtosisCara miotónicaAnomalías del conducto lacrimalRetraso de la erupción dental | Nariz con raíz amplia y punta bulbosaMejillas prominentesMicro/retrognatia o prognatismoAnomalías dentales |
| Endocrinológicos | HipotiroidismoRetraso ponderoestatural | ||
| Respiratorios | Laringomalacia |
Las mutaciones mayoritariamente reportadas fueron del exón 18, hallándose proximales en SGP y distales en SBBYSS; confiriendo mayor gravedad cuanto más proximal2.
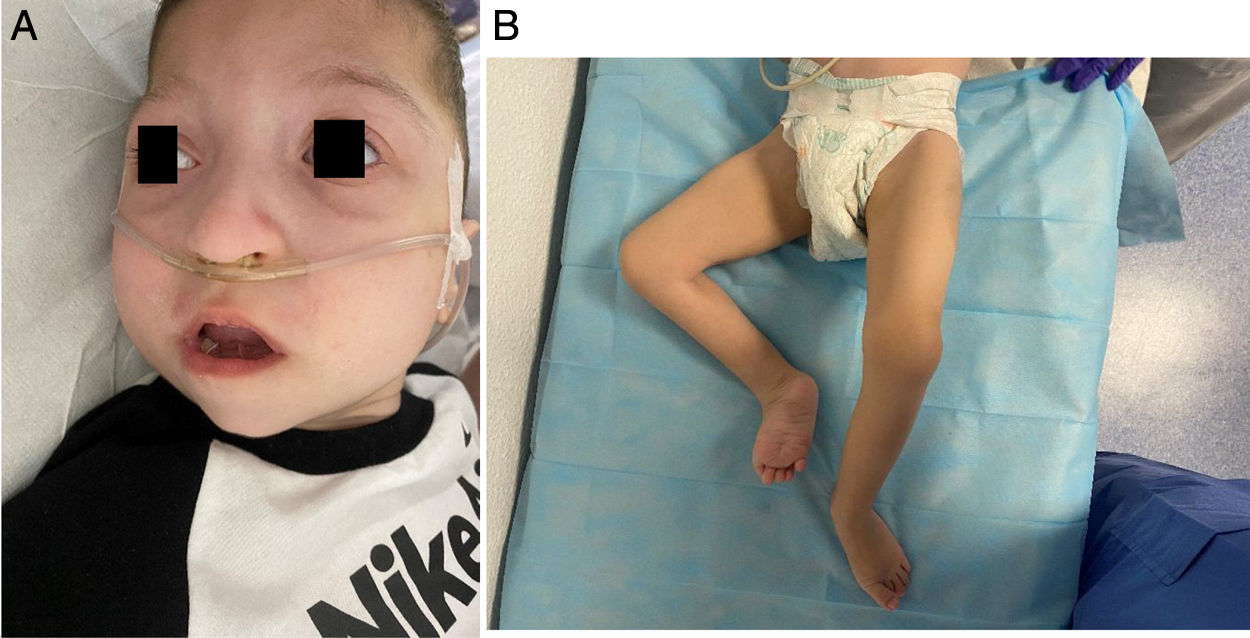
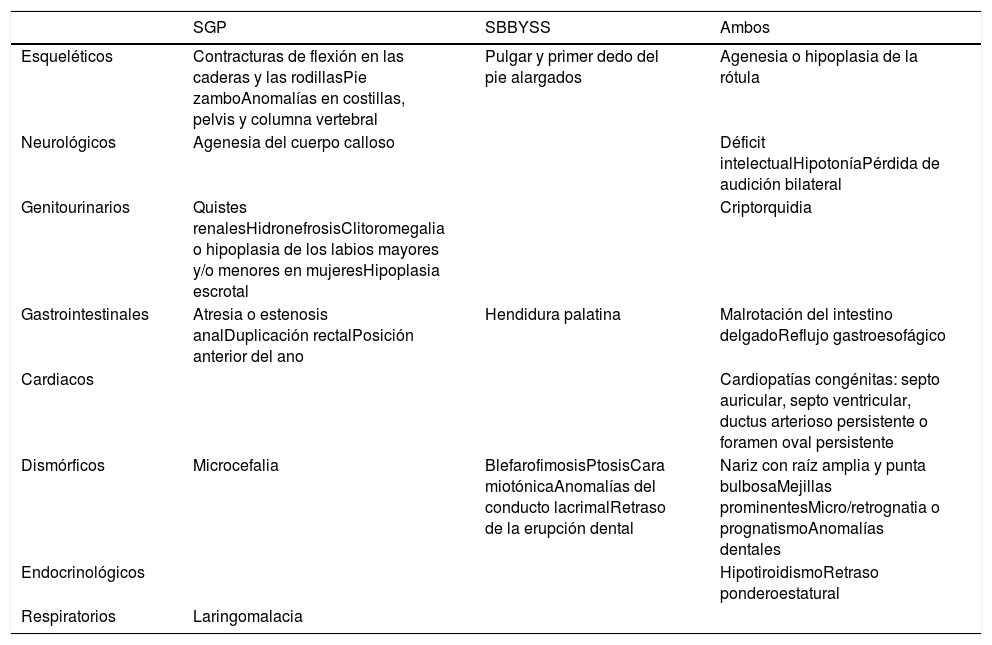
Paciente de 2 años, quinto de padres sanos, no consanguíneos. Antecedentes perinatales/familiares no relevantes. Presenta facies característica con microcefalia (fig. 1A), displasia de caderas con luxación congénita izquierda, criptorquidia bilateral, hipospadias y agenesia e hipoplasia rotulianas derecha e izquierda, respectivamente (fig. 1B). Neurológicamente, hipotonía axial y retraso psicomotor con disgenesia del cuerpo calloso y retraso de la mielinización. Cardiorrespiratoriamente, hipoxemia oxigenodependiente, traqueomalacia y miocardiopatía hipertrófica no obstructiva. Endocrinológicamente, hipertirotropinema. Gastrointestinalmente, gastrostomía por disfagia y malnutrición crónica e ileostomía por obstrucción intestinal al año de vida.

A. Cara miotónica, hendidura palpebral alargada, telecanto, proptosis, orejas de implantación baja, puente nasal bajo, punta nasal bulbosa, labio superior fino, filtrum alargado, microretrognatia y mejillas prominentes. B. Pies equinovaros, pulgares gordos y dedos largos, hallux valgus con segundo dedo infraducto.
Realizamos secuenciación NGS del exoma dirigido a síndrome de Charge y genes relacionados cubriendo el 99,47% de las regiones codificantes. Detectamos, al inicio del exón 18, la variante KAT6B:c.4090G>T;(p.Glu1364Ter) de novo, no descrita en la literatura, clasificada por predictores (MutationTaster) como deletérea con frecuencia 0,00 en controles.
El paciente presenta un fenotipo grave descrito en las mutaciones más proximales del exón 18 y nuestros hallazgos concuerdan con lo ya descrito en la literatura, en donde se defiende la denominación de «trastornos del espectro KAT6B»3, siendo la distinción clínica cada vez más obsoleta y la genotípica, el futuro.

