

Caso clínico
Niña de 4 años que ingresó por presentar durante el sueño nocturno un cuadro agudo de tos seguido de náuseas y vómitos persistentes, palidez de piel, desviación mantenida de la mirada hacia la derecha y atenuación del nivel de conciencia. No presentaba movimientos anormales y el tono muscular estaba disminuido. El episodio duró de 10 a 15 min quedando dormida y permaneciendo asintomática tras despertar.
No constaban antecedentes patológicos personales ni familiares de interés y su desarrollo neurológico fue normal. Seis meses antes la paciente había presentado un cuadro de morfología y duración muy similar, también durante el sueño, aunque esta vez sin tos. Posteriormente presentó somnolencia e hiporreactividad, con buena recuperación en la hora siguiente. En esta ocasión tampoco aparecieron movimientos tonicoclónicos. El líquido cefalorraquídeo (LCR) y la tomografía computarizada (TC) craneal fueron normales. Un electroencefalograma (EEG) en vigilia, efectuado a las 4 h del episodio, mostraba lentificación sobre región temporal derecha sin anomalías paroxísticas asociadas. En ambos eventos el proceso generó una enorme angustia en los padres. Ninguno de los dos episodios se asoció a fiebre ni a otros procesos intercurrentes. Tampoco coinciden con la administración de ningún fármaco.
En la exploración física presentaba un buen estado general, de nutrición y desarrollo. Estaba afebril. La auscultación cardiopulmonar y abdominal fue normal. Neurológicamente estaba consciente y orientada. Algo asustada e irritable. Fuerza, tono y motilidad normal. Pupilas isocóricas y normorreactivas. No había rigidez de nuca ni otros signos meníngeos. Fondo de ojo normal.
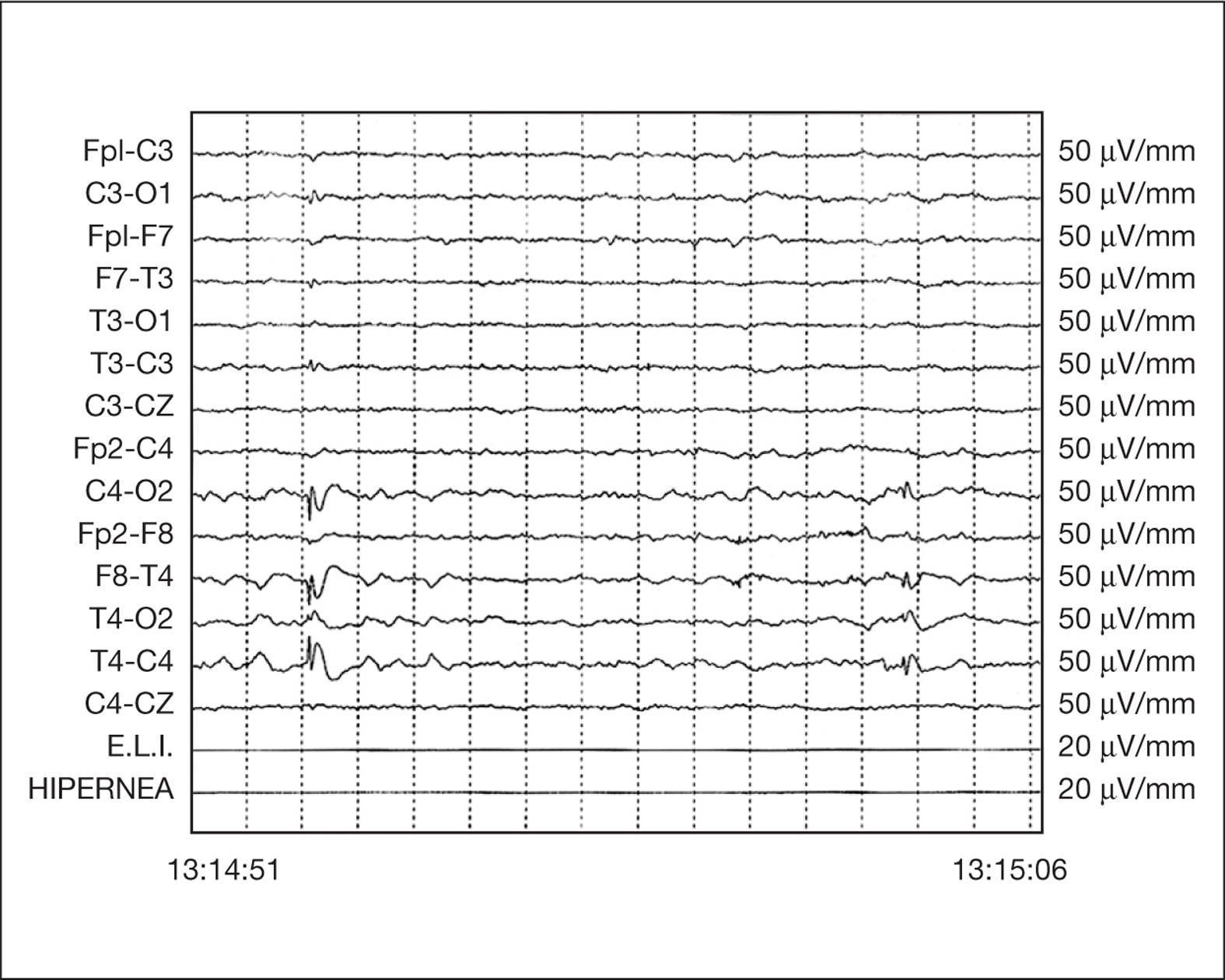
Entre las exploraciones complementarias el hemograma, bioquímica e iones en sangre fueron normales, así como el pH y la gasometría venosa, fracciones de hemoglobina y tóxicos en orina. Un EEG a las 24 h del evento mostraba, sobre una actividad basal normal y bien estructurada, la presencia de complejos punta-onda repetidos de elevada amplitud sobre región temporooccipital derecha, que se bloqueaban con la apertura de párpados (fig. 1).
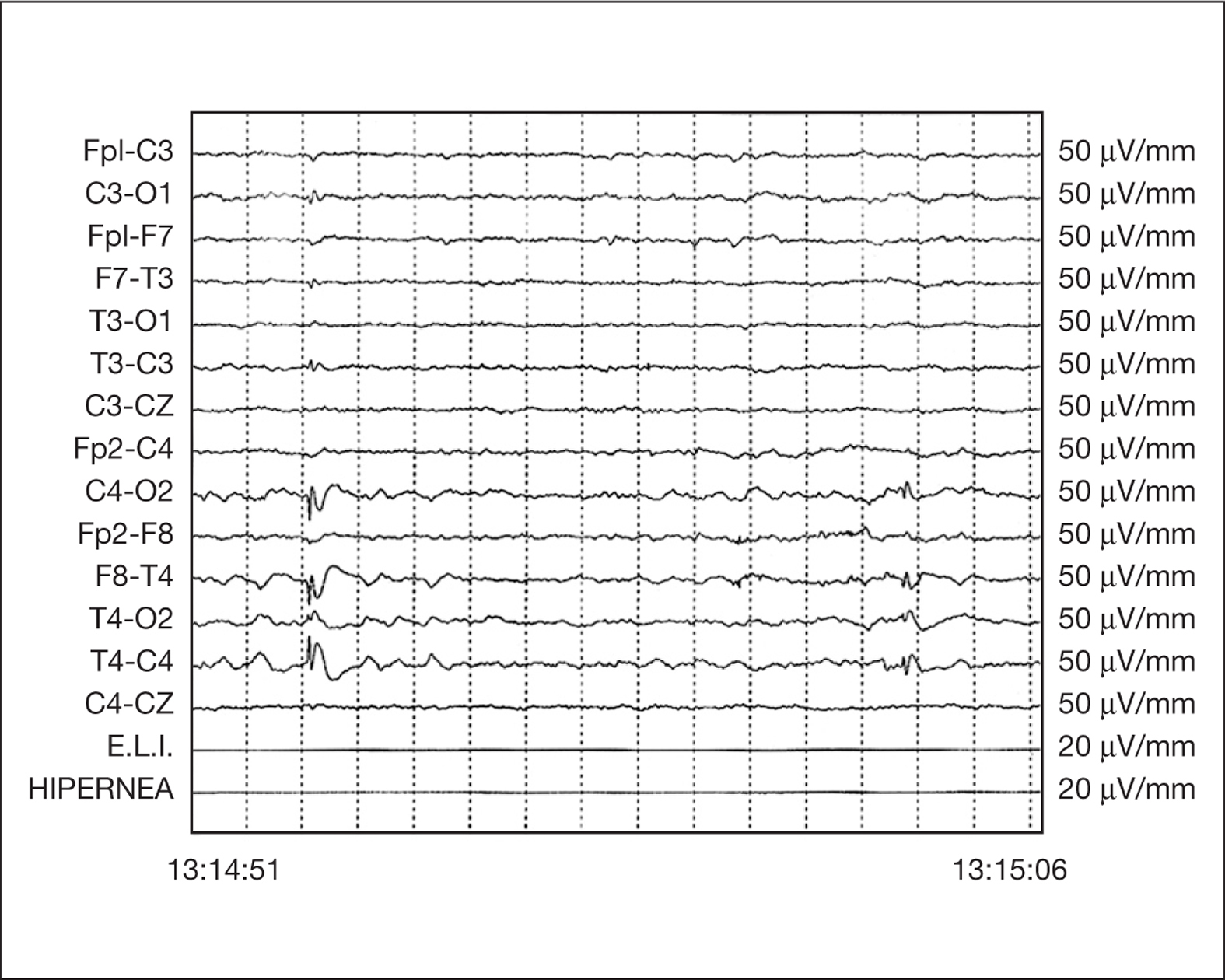
Figura 1. Trazado EEG intercrítico en vigilia. Paroxismos repetidos de punta seguida de onda lenta de elevada amplitud (más de 300 mV) sobre región temporooccipital derecha.
Pregunta
¿Cuál es su diagnóstico?
Crisis epilépticas autonómicas. Síndrome de panayiotopoulos
Las crisis autonómicas y el estado epiléptico autonómico, característicos de la epilepsia occipital benigna de la infancia de inicio temprano (síndrome de Panayiotopoulos)1,2, son episodios con frecuencia infradiagnosticados y confundidos habitualmente con episodios no epilépticos, como gastroenteritis, migraña atípica, encefalitis, hemorragia cerebral o síncopes cardiogénicos1,3.
El síndrome de Panayiotopoulos es un tipo bien definido, no infrecuente y autolimitado, de epilepsia idiopática benigna en la infancia, oficialmente reconocido por la Liga Internacional contra la Epilepsia, aunque erróneamente equiparado con la epilepsia occipital4. Aunque previamente se haya considerado excepcional, actualmente se estima una prevalencia del 13 % en niños entre 3-6 años con una o más convulsión no febril, o del 6 % en el grupo de 1-15 años1,2,5. Afecta a pacientes de ambos sexos, previamente sanos, siendo la edad de inicio entre 1-12 años (pico máximo entre los 3-6 años, media 5 años)1,5-10.
La presentación clínica típica es un niño de 3-6 años, consciente, capaz de hablar, que se queja de sensación de malestar, vomita repetidamente y presenta palidez cutaneomucosa. Dos tercios de las crisis son de predominio nocturno. Durante ellas, los pacientes presentan una combinación variable de síntomas que pueden incluir cambios autonómicos, alteraciones en su capacidad de interacción con el ambiente o el comportamiento, vómitos, desviación tónica de los ojos y reactividad disminuida (alteración o pérdida de conciencia). A veces progresa hacia una hemiconvulsión con marcha jacksoniana (19 %) o crisis generalizadas (21 %)1,7-10. El vómito crítico, presente en el 60-100 % de los casos, puede ocurrir en cualquier momento durante el episodio, aunque con frecuencia lo hace al inicio. La desviación lateral conjugada de los ojos puede acompañarse de desviación cefálica que aparece en el 60 % de las crisis. Otras manifestaciones autonómicas pueden aparecer a la vez o a lo largo de la crisis; entre ellas se incluyen palidez, y con menor frecuencia tos, cianosis o enrojecimiento facial, midriasis, miosis, alteraciones cardiorrespiratorias, incontinencia esfinteriana, hipersalivación y cambios en la motilidad intestinal. Las crisis autonómicas tienden a ser de larga duración y el 44 % de ellas se prolongan más de 30 min (estado epiléptico autonómico). La duración del 56 % restante varía entre 1 y 30 min con una media de 9 min1,2,5,8-10.
El EEG intercrítico está alterado en el 90 % de los pacientes y los hallazgos pueden ser variables1,2,6-10. Dos tercios (68 %) de los pacientes presentan al menos un EEG con paroxismos occipitales de punta onda, con o sin generalización, tal como ocurrió en nuestro caso, y que a menudo (64 %) coinciden con otras descargas extraoccipitales. El tercio restante (32 %) nunca muestra paroxismos occipitales, pudiendo presentar paroxismos extraoccipitales exclusivamente (21 %), EEG normal (9 %) o descargas generalizadas (2 %)1,5,7-10. En ocasiones el EEG puede ser normal tras el episodio crítico, tal como ocurrió en el primero de los episodios de nuestro paciente.
La fisiopatología del síndrome de Panayiotopoulos es desconocida, pero es probable que se deba a una activación epileptiforme del centro del vómito y del hipotálamo1. Por ello, el síndrome de Panayiotopoulos no se debe considerar como un tipo de epilepsia idiopática occipital, como con frecuencia es clasificado4.
El pronóstico es bueno. El 27 % de los niños presentan sólo una crisis. La mitad (47 %) presenta de 2 a 5 episodios y sólo el 5 % más de 10 episodios. Una pequeña proporción (21 %) presentan crisis rolándicas durante la fase activa del síndrome, o evolutivamente desarrollan una epilepsia rolándica benigna1,5,9. Actualmente se recomienda no tratar farmacológicamente a estos niños, salvo crisis repetidas en cuyo caso se utiliza la carbamazepina1,8. La remisión a menudo ocurre 1-2 años después del inicio del tratamiento.
El conocimiento de este síndrome por parte del pediatra, en muchas ocasiones dramático por su sintomatología3, puede ayudar a un reconocimiento precoz de los síntomas y a una optimización del diagnóstico y tratamiento.

