

La asfixia intraparto es una de las causas más frecuentes de muerte neonatal precoz pero también puede, en los supervivientes, evolucionar a una encefalopatía hipóxico-isquémica responsable de una elevada morbilidad neurológica. La presencia de episodios de hipoxia-isquemia prolongados conduce a un rápido agotamiento energético en los tejidos exclusivamente dependientes del metabolismo aeróbico, como el sistema nervioso central. El déficit energético conlleva una paralización de las bombas ATP-dependientes y subsiguiente pérdida del potencial neuronal transmembrana. La población neuronal de las regiones más sensibles del SNC mueren por necrosis, mientras que en otras áreas se produce una hiperexcitabilidad neuronal con entrada masiva de calcio iónico, activación de NO-sintasa, generación de radicales libres que alteran el funcionamiento mitocondrial, provocando un fallo energético secundario y muerte neuronal por apoptosis. Recientemente se ha propuesto una tercera fase en la que factores como la inflamación persistente y los cambios epigenéticos causarían un bloqueo de la maduración de los oligodendrocitos, alteración de la neurogénesis, del crecimiento axonal y de la sinaptogénesis. En este contexto, el estrés oxidativo va a tener un papel protagonista como responsable tanto en causar daño directo al SNC como en activar cascadas metabólicas conducentes a la apoptosis e inflamación. La hipotermia moderada precoz, al preservar las reservas energéticas y disminuir la formación de especies reactivas de oxígeno, atenuará el daño cerebral posreanimación. La combinación de la hipotermia con terapias coadyuvantes para modular el estrés oxidativo podría contribuir a mejorar el pronóstico.
Birth asphyxia is one of the principal causes of early neonatal death. In survivors it may evolve to hypoxic-ischaemic encephalopathy and major long-term neurological morbidity. Prolonged and intense asphyxia will lead to energy exhaustion in tissues exclusively dependent on aerobic metabolism, such as the central nervous system. Energy deficit leads to ATP-dependent pumps blockage, with the subsequent loss of neuronal transmembrane potential. The most sensitive areas of the brain will die due to necrosis. In more resistant areas, neuronal hyper-excitability, massive entrance of ionic calcium, activation of NO-synthase, free radical generation, and alteration in mitochondrial metabolism will lead to a secondary energy failure and programmed neuronal death by means of the activation of the caspase pathways. A third phase has recently been described that includes persistent inflammation and epigenetic changes that would lead to a blockage of oligodendrocyte maturation, alteration of neurogenesis, axonal maturation, and synaptogenesis. In this scenario, oxidative stress plays a critical role causing direct damage to the central nervous system and activating metabolic cascades leading to apoptosis and inflammation. Moderate whole body hypothermia to preserve energy stores and to reduce the formation of oxygen reactive species attenuates the mechanisms that lead to the amplification of cerebral damage upon resuscitation. The combination of hypothermia with coadjuvant therapies may contribute to improve the prognosis.
Los tejidos llamados oxirreguladores representados por el sistema nervioso central (SNC) y el miocardio precisan una elevada cantidad de energía para el mantenimiento de los potenciales de transmembrana y, por ello, dependen del metabolismo aeróbico que genera energía mucho más eficientemente que el anaeróbico1. En el SNC la transmisión del impulso nervioso precisa del concurso de las bombas iónicas ATP-asa dependientes, que consumen una elevada cantidad de energía. Además, el SNC es incapaz de acumular reservas energéticas en forma de sustratos de acción rápida como fosfocreatina o glucógeno, y depende exclusivamente de un aporte continuado de glucosa/oxígeno. Por lo tanto, la privación de oxígeno y glucosa conducen a un rápido agotamiento de las reservas energéticas y a la muerte celular en pocos minutos2–4.
La asfixia intraparto se caracteriza por periodos de hipoxia/isquemia durante el trabajo del parto que, dependiendo de su intensidad, pueden ocasionar la muerte o evolucionar a una encefalopatía hipóxico-isquémica (EHI). La mortalidad por EHI en países industrializados es de 1-8 muertes/1.000 nacidos vivos, pudiendo llegar en países no industrializados a cifras de 26 muertes/1.000 nacidos vivos. El reto de la neonatología es lograr disminuir la morbimortalidad en la EHI, ya que, a pesar de la generalización de la hipotermia, aún fallecen el 45% de los pacientes, y entre los que sobreviven un porcentaje elevado padecerá discapacidades graves5.
Este artículo de revisión tiene como objetivo describir las bases bioquímicas del metabolismo oxidativo y efectuar una descripción de las terapias coadyuvantes que pueden contribuir a regular la generación de radicales libres y mejorar los resultados de la hipotermia.
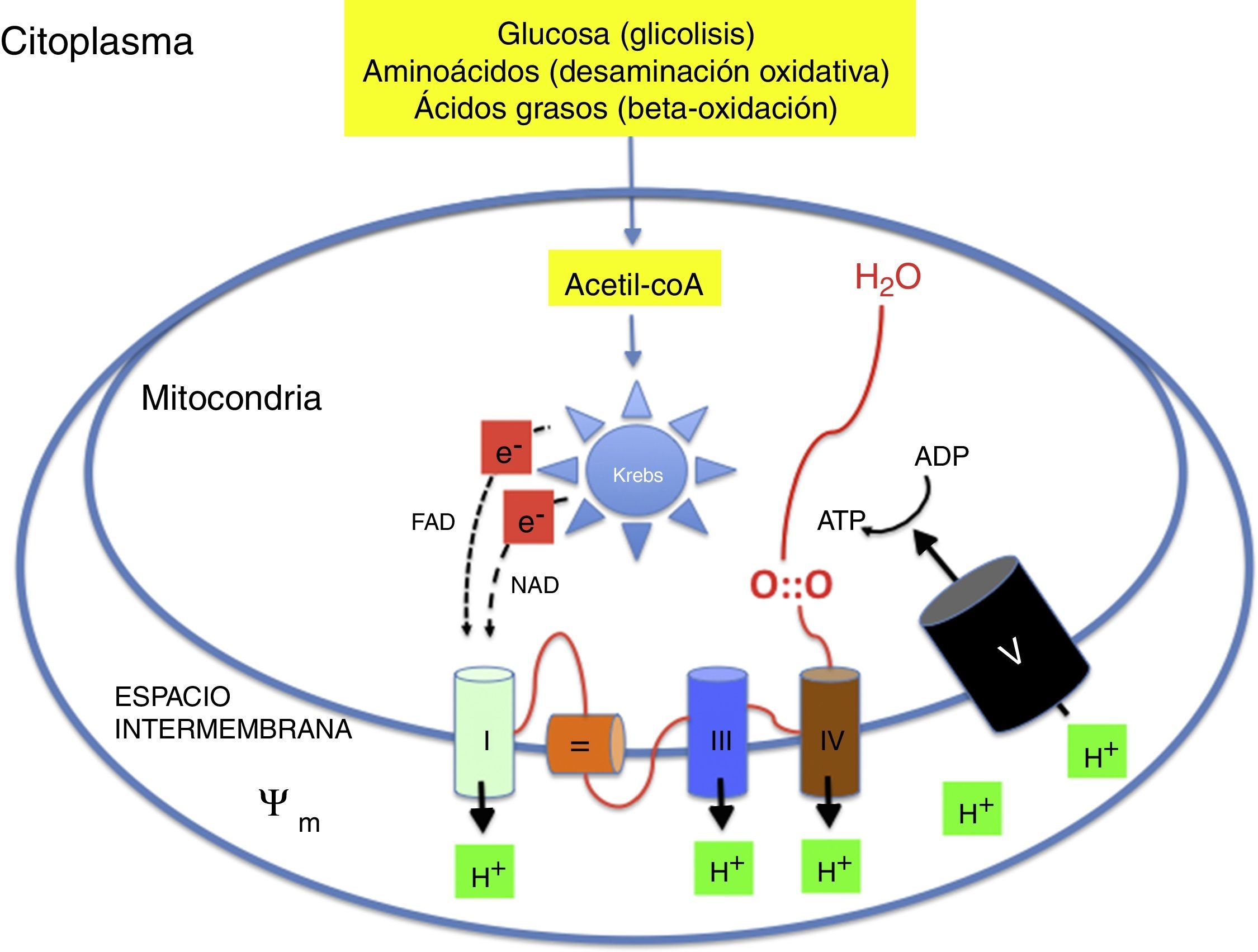
Metabolismo oxidativo2,6–9Metabolismo aeróbico y fosforilación oxidativa (fig. 1)La energía necesaria para la vida de los seres multicelulares requiere el concurso del oxígeno en la fosforilación oxidativa mitocondrial. Sustratos como glucosa, aminoácidos y ácidos grasos son transformados en acetil-coenzima A (acetil-coA) en la matriz de las mitocondrias. La acetil-coA es metabolizada por los distintos componentes del ciclo de Krebs. Durante este proceso se liberan electrones con un alto contenido energético que son transportados por nicotín-adenín-dinucleótido y flavín-adenín-dinucleótido hasta la cadena respiratoria. La cadena respiratoria está formada por una serie complejos enzimáticos (i, iii y iv) alineados capaces de extraer protones (H+) hasta el espacio intermembranoso generando un potencial de transmembrana (ψm). La ATP-sintasa introduce de nuevo los protones en el interior de la mitocondria a favor de ψm liberando energía que se utiliza para sintetizar ATP a partir del ADP. El oxígeno capta los electrones procedentes del ciclo de Krebs evitando que dañen las estructuras mitocondriales. El aprovechamiento energético es tal que de una molécula de glucosa se generan 36 molecules de ATP y de una molécula de ácido palmítica 106 moleculas de ATP2,6–9.

Mecanismo de la fosforilación oxidativa mitocondrial. Los nutrientes esenciales son transformados metabólicamente en acetil-coA y liberan su energía en forma de electrones de alta energía que son transportados a la cadena respiratoria. La energía se utiliza para generar un potencial de transmembrana que se utiliza en la resíntesis de ATP.
La reducción del oxígeno se completa con la incorporación de 4 electrones a su orbital más externo. La reactividad del oxígeno es limitada, de modo que se reduce secuencialmente (reducción lenta). Sin embargo, en presencia de metales de transición como cobre o hierro la reactividad aumenta exponencialmente (reducción rápida). La presencia de abundante hierro en el SNC incrementará, por lo tanto, los procesos oxidativos, como ocurre en la EHI. En el modo secuencial el oxígeno es reducido por un electrón a anión superóxido (•O2−), por dos electrones a peróxido de hidrógeno (H2O2), o por tres electrones a radical hidroxilo (•OH). Todos estos compuestos que se generan de forma habitual durante el proceso de la fosforilación oxidativa mitocondrial se conocen como especies reactivas de oxígeno. Sin embargo, algunas de estas especies, como el anión superóxido o el radical hidroxilo, y otros más como el óxido nítrico (•NO) o los peroxilos lipídicos (LOO•) son radicales libres, es decir, son especies reactivas que tienen en su orbital más externo un electrón no apareado que les confiere una gran reactividad reaccionando con sustratos próximos para arrebatarle ese electrón que precisan para su estabilidad, y generando así una reacción en cadena que pueden dañar estructuras celulares. El radical libre más abundante en nuestra biología es el anión superóxido. No todas las especies reactivas son radicales libres; así, el peróxido de hidrógeno (H2O2), aunque actúa como señalizador celular para numerosas funciones biológicas, puede ser un precursor de radicales libres, especialmente cuando hay presentes metales de transición (Fe++/Fe+++; Cu+)6.
.")
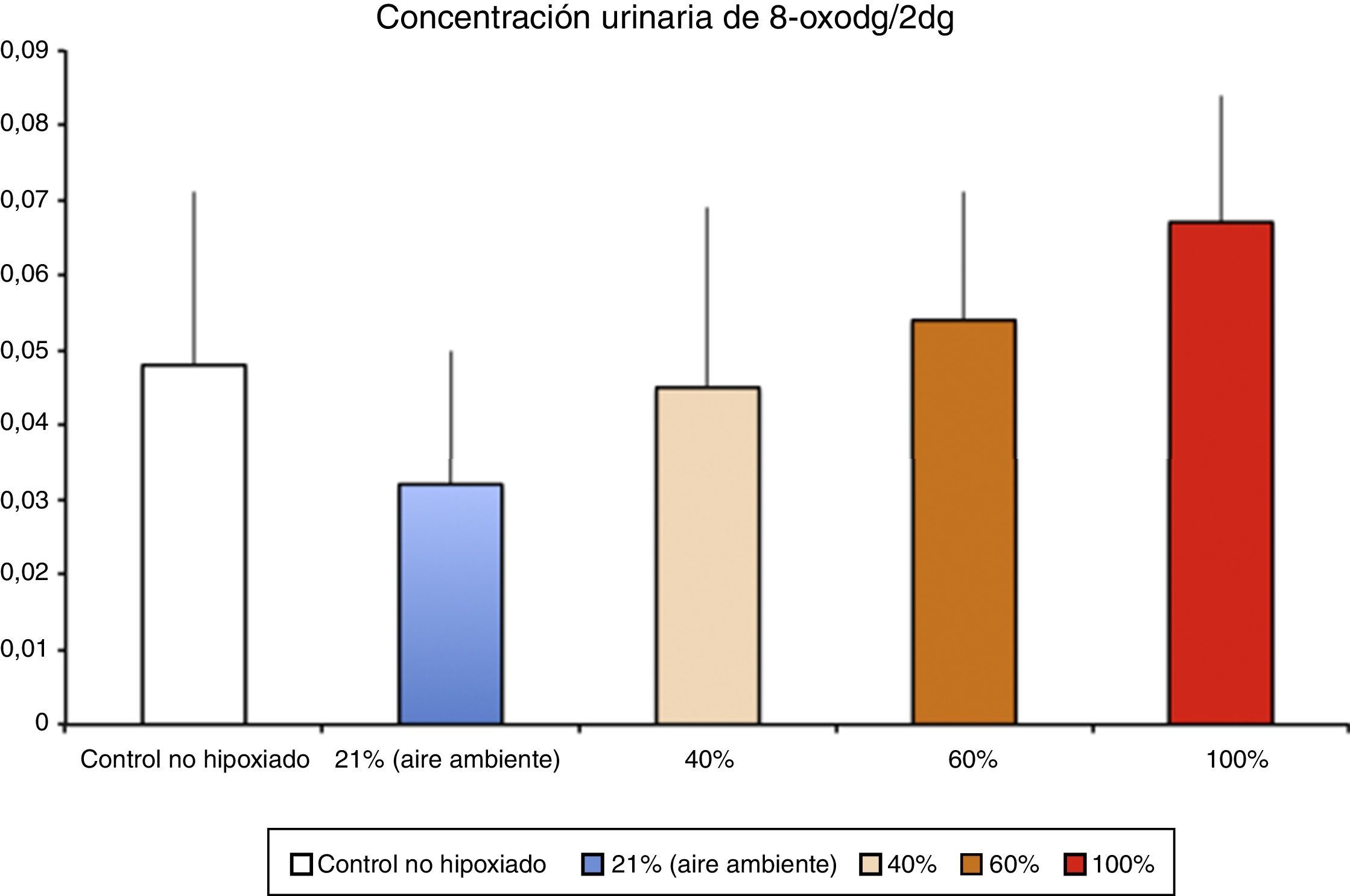
Los niveles de biomarcadores relacionados con daño al ADN en un modelo de hipoxia/reoxigenación de cerdos recién nacidos correlacionaron con la fracción inspiratoria utilizada durante la reoxigenación (Solberg et al.17).
La principal fuente fisiológica de radicales libres es la actividad mitocondrial. En la mitocondria el oxígeno se reduce completamente y, al reaccionar con protones (H+) da lugar a la formación de agua. En situaciones de estrés metabólico (reanimación, infección, nutrición parenteral, etc.) se produce un incremento en la formación de radicales libres que puede causar desestructuración de las mitocondrias y bloquear la generación de energía. Fuente importante de estrés oxidativo son la activación del sistema NADPH-oxidasa (NOX) en los fagocitos como respuesta a la infección, suplementación con oxígeno o administración de ciertos fármacos6,8,9.
Sistemas antioxidantes biológicosEl estrés oxidativo refleja una situación de desequilibrio entre la formación de radicales libres y su neutralización. Un cierto grado de estrés oxidativo es necesario para el funcionamiento de sistemas enzimáticos que precisan de las especies reactivas de oxígeno para la señalización de sus funciones celulares. Sin embargo, en situaciones de profundo desequilibrio se produce daño tisular conducente a patología aguda y crónica9.
El sistema de defensa antioxidante (SDA) incluye mecanismos enzimáticos y no enzimáticos. Las enzimas antioxidantes eliminan las especies reactivas mediante reacciones químicas. Los SDA no enzimáticos están constituidos por proteínas que fijan a metales de transición (transferrina, ceruloplasmina, ferritina), vitaminas que bloquean la peroxidación lipídica (A, E y C) o compuestos de bajo peso moleculares que reducen las especies reactivas. El glutatión, en su forma reducida (GSH), se une a una segunda molécula idéntica para formar el glutatión oxidado (GSSG), cediendo dos electrones a un radical libre y contribuyendo así a su normalización. El GSSG por acción de GSH-reductasa vuelve a su forma reducida. Entre las enzimas antioxidantes más relevantes están las superóxido dismutasas (SOD), que catalizan la conversión de anión superóxido en peróxido de hidrógeno; catalasas (CAT) y glutatión peroxidasas (GPx) que transforman el peróxido de hidrógeno en agua y oxígeno. Otros sistemas incluyen glutarredoxinas, tiorredoxinas, peroxirredoxina, y las heme-oxigenasas que eliminan el complejo heme pro-oxidante de los glóbulos rojos y lo transforman en biliverdina. El SDA se desarrolla tardíamente en la gestación con la finalidad de preparar al feto para la oxigenación posnatal6,9.
Estrés y daño oxidativo y su mediciónLos radicales libres provocan alteraciones en proteínas, lípidos, glúcidos, ARN y ADN. Hay marcadores globales, como el cociente glutatión reducido/oxidado (GSH/GSSG). Cuando hay una situación pro-oxidante, GSSG aumenta y el cociente disminuye, y lo contrario ocurre si hay un estado antioxidante o reductor, con aumento del GSH. Otro modo es medir la actividad de las enzimas antioxidantes. Así, frente a una mayor agresión oxidativa habrá un incremento proporcional de la actividad enzimática como respuesta protectora. Sin embargo, en la actualidad se están buscando biomarcadores que puedan detectar en fluidos biológicos los derivados de la oxidación de distintos componentes celulares. En años recientes se han validado una serie de marcadores de daño oxidativo en los distintos biofluidos en el período neonatal10–16.
Fisiopatología de la isquemia reperfusiónGeneración de radicales libres de oxígeno en la isquemia reoxigenaciónLa hipoxia/isquemia ocasiona una depleción de ATP que provoca una disfunción inicialmente reversible, pero si se prolonga puede llegar a ser irreversible. Los tejidos llamados oxirreguladores, cuyo paradigma es el SNC, son los más sensibles al déficit de oxigenación. Los estudios realizados en los años setenta demostraron que durante la reperfusión/reoxigenación después de un período hipóxico-isquémico se generaba una amplificación notable del daño inicial. La gravedad del daño por reoxigenación estaba en relación directa con el tiempo y la intensidad de la hipoxia-isquemia así como de la concentración de oxígeno utilizada durante la reoxigenación/reperfusión8. En un modelo experimental de hipoxia-isquemia en cerdos recién nacidos, las lesiones histológicas del SNC y la eliminación de marcadores de daño a ADN en orina (fig. 3) fueron proporcionales a la FiO2 utilizada durante la reoxigenación17. En la actualidad se están estudiando marcadores específicos de hipoxia tisular. El lactato refleja con fiabilidad la intensidad de la hipoxia pero no refleja la duración de la hipoxia tisular. En un modelo experimental de cerdo recién nacido se ha demostrado que la suma combinada de metabolitos derivados de la oxidación de purinas, pirimidinas y fosfolípidos obtenidos por espectrometría de masas da lugar a un «score metabólico» fiable y reproducible para determinar la intensidad y la duración de la hipoxia cerebral y proporcionar un pronóstico o facilitar la decisión sobre la terapia más adecuada18–20.

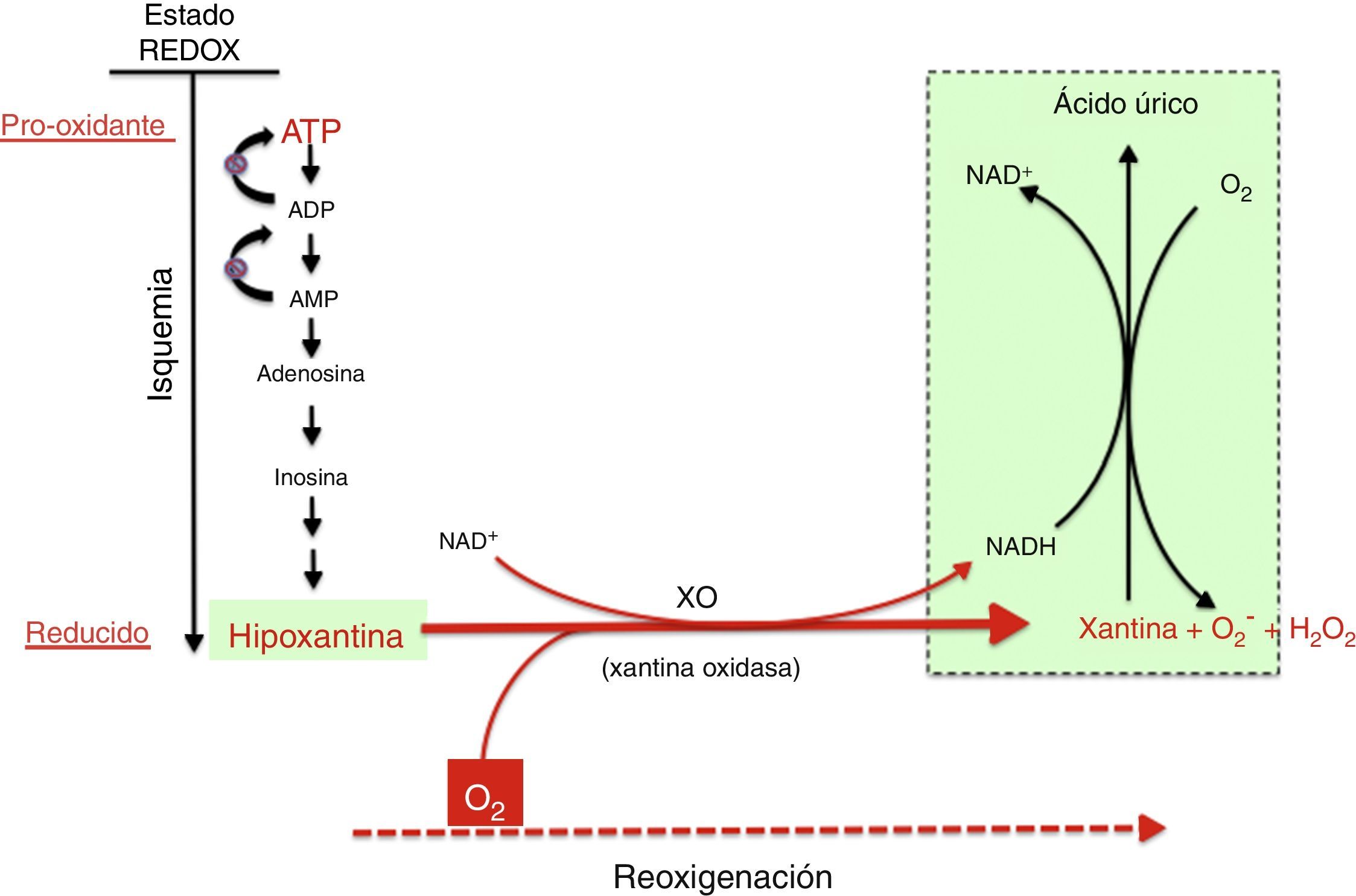
Mecanismo de generación de radicales libres por acción del complejo xantina óxido-reductasa. Durante la hipoxia prolongada se acumulan derivados purínicos del ATP especialmente hipoxantina. En la reoxigenación, la xantina reductasa se transforma en xantina oxidasa, que generará una elevada cantidad de especies reactivas de oxígeno, provocando un estrés oxidativo.
Las fuentes de especies reactivas durante la isquemia-reoxigenación (IR) son múltiples, siendo las más relevantes el complejo respiratorio mitocondrial y el sistema de la xantina oxidasa (XO). También se han implicado, aunque en menor medida, el sistema NADPH oxidasa (NOX) y el desacoplamiento de la NO-sintasa8.
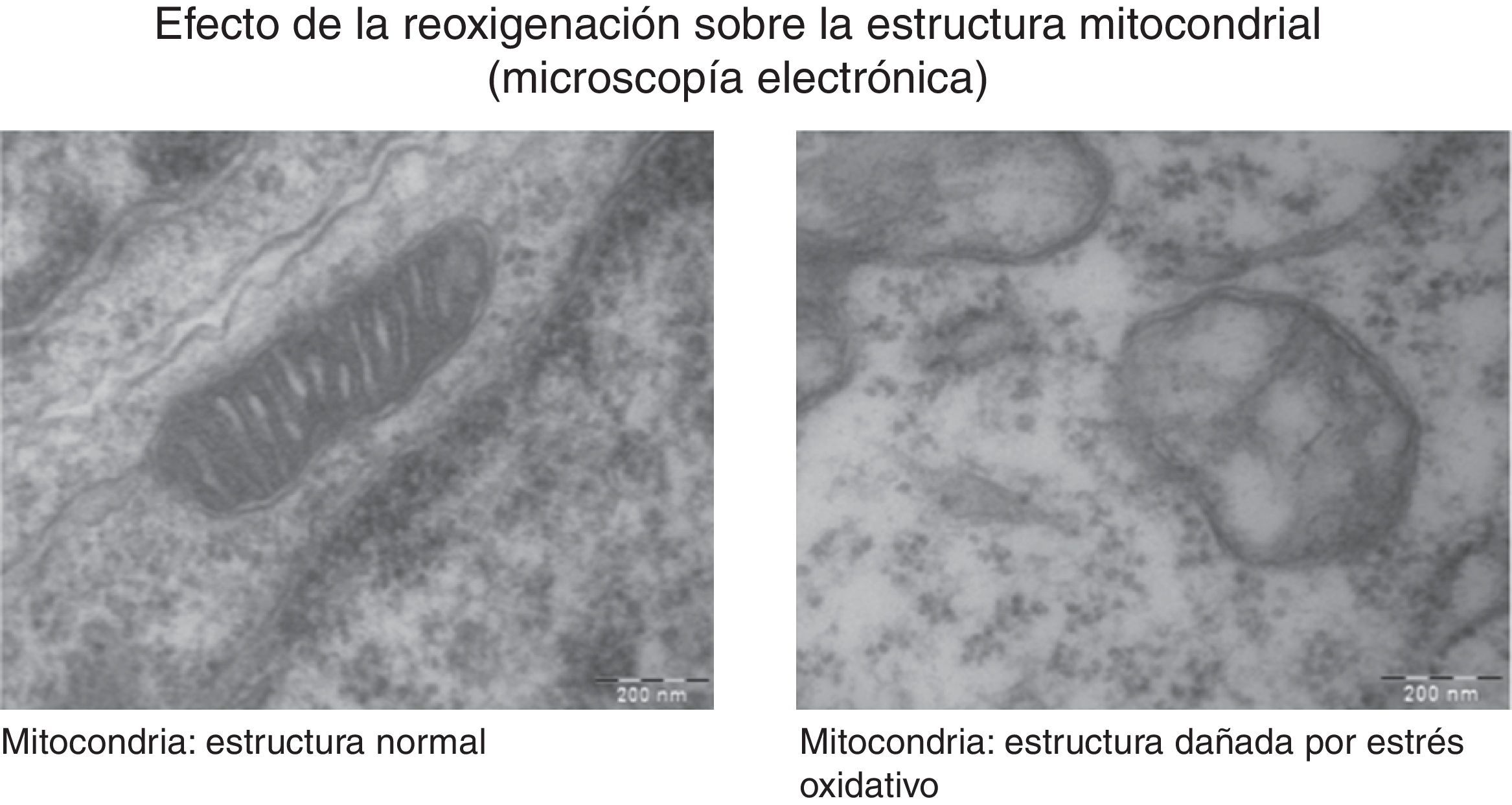
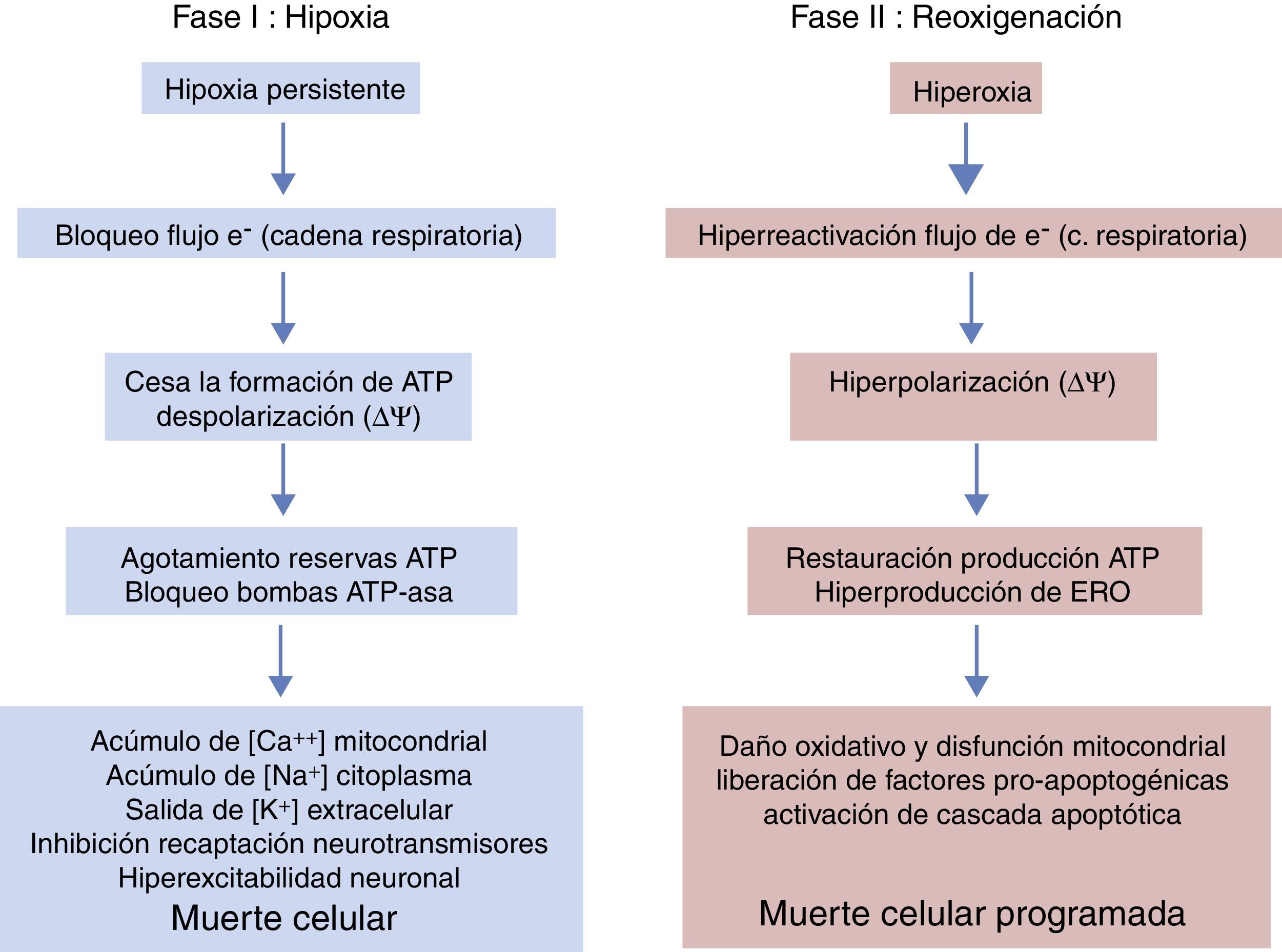
La generación de radicales libres en condiciones normales está regulada. Los electrones que fluyen a través de la cadena respiratoria son captados casi en su totalidad (98%) por el oxígeno, generándose tan solo un 2% de especies reactivas. Sin embargo, en situaciones de hipoxia la falta de oxígeno provoca un bloqueo de la circulación de electrones en la cadena respiratoria y un descenso del Ψm. Niveles muy bajos del Ψm inhiben la actividad ATP-sintasa, se detiene la producción de ATP, se produce una lisis mitocondrial y la muerte celular. Durante la reoxigenación se recupera el Ψm y se reinicia la síntesis de ATP. Sin embargo, la brusca reactivación del flujo de electrones ocasiona la generación excesiva de especies reactivas que pueden amplificar la lesión neuronal inicial por apoptosis2,7,8. En la figura 4 se muestran estudios de microscopia electrónica en cortes de cerebro de un modelo experimental de ratón sometido a hipoxia/reoxigenación en los que se ha captado la desestructuración, la edematización, la pérdida de las crestas, la rotura de las membranas y finalmente la vacuolización y la destrucción de las mitocondrias.
.")
Las imágenes muestran las variaciones morfológicas y estructurales que incluyen desestructuración, edematización, pérdida de las crestas, rotura de las membranas y finalmente vacuolización y destrucción de las mitocondrias cerebrales en un modelo de ratón recién nacido sometido a distintas concentraciones de oxígeno. Cortesía de Isabel Torres-Cuevas (tesis doctoral; director: Máximo Vento).
En la fase de reoxigenación de la IR, el oxígeno reacciona con la hipoxantina y la XO generando un brote de radicales superóxido y peróxido de hidrógeno que, a su vez, darán lugar a otras especies reactivas en cadena amplificando el daño tisular inicial producido por la isquemia prolongada. La actividad de XO está también regulada a nivel postraslacional por la presión parcial de oxígeno existente en los tejidos de modo que, cuando menor es la presión de oxígeno, mayor es la actividad de la XO. Ello explicaría que el daño por estrés oxidativo ya se puede iniciar en la fase de hipoxemia y se ve amplificado en la de reoxigenación. Recientemente se ha demostrado la capacidad de la XO de generar •NO a partir de nitritos. La generación simultánea de superóxido y de •NO daría lugar a la formación de peroxinitrito, un radical nitrosativo de gran agresividad21,22.
Períodos evolutivos de la encefalopatía hipóxico-isquémicaLa evolución de la EHI se divide en 3 fases (fig. 5), aunque en realidad sea un continuum en el que puede haber interferencias entre las mismas. La primera fase tiene una duración de 6h y se caracteriza por una disminución del flujo sanguíneo al feto causando una hipotensión arterial sistémica y pérdida de la autorregulación en la circulación cerebral. La isquemia cerebral produce hipoxia, acidosis y daño cerebral como consecuencia del fallo energético primario. La falta de oxígeno se suple mediante el metabolismo anaeróbico causando acidosis láctica, depleción de ATP, acúmulación de Na+, Ca++, agua e inhibición de la recaptación de neurotransmisores con hiperexcitabilidad neuronal secundaria. La entrada masiva de Ca++ intracelular produce una activación de lipasas, de NO-sintasa, producción de radicales libres, disfunción mitocondrial y liberación de sustancias apoptogénicas. En una segunda fase (6-48h) se mantiene el estado de excitotoxicidad, empeora la función mitocondrial y el estrés oxidativo secundario a alteraciones del potencial de membrana con una reducción de la síntesis de ATP en un entorno de alcalosis intracelular a pesar de la adecuada oxigenación. Finalmente, hay una tercera fase, que puede durar días, semanas e incluso meses, caracterizada por la presencia de inflamación y cambios epigenéticos que conducen a alteraciones en el crecimiento axonal, neurogénesis y sinaptogénesis23,24.
Estrategias frente al daño causado por especies reactivas de oxígeno
La prevención de la agresión producida por especies reactivas de oxígeno requiere una intervención muy precoz, ya que esta se puede haber iniciado intraútero y se reactiva durante la reanimación posnatal. Por lo tanto, dentro del esquema de tratamiento serían intervenciones indicadas en la fase de fallo energético primario. Podrían ser realizadas o bien en la gestante durante el trabajo de parto cuando se apreciase pérdida importante del bienestar fetal, o en el recién nacido con datos clínicos y/o electrofisiológicos de EHI en los primeros minutos de vida25,26.
A continuación se describen brevemente las intervenciones relacionadas con aspectos patogénicos del estrés oxidativo.
HipotermiaLa hipotermia produce un descenso de la actividad metabólica de tejidos como el SNC con la consiguiente reducción de las necesidades de intercambio iónico y del consumo de ATP. El descenso en el ritmo de la actividad mitocondrial y el acoplamiento de las proteínas desacopladoras (UCP) reduce significativamente la generación de especies reactivas sin pérdida de los potenciales de transmembrana y bloqueando la liberación de proteínas apoptogénicas. Los mecanismos de acción a nivel celular y la eficacia clínica de la hipotermia han sido ampliamente investigados. A pesar de la eficacia contrastada de este tratamiento como estrategia neuroprotectora en EHI moderada y grave, hasta el 45% de los pacientes tratados, especialmente aquellos con EHI grave, presentan pronóstico adverso. Esto podría cambiar en un futuro inmediato gracias a los estudios sobre terapias coadyuvantes que se están realizando a nivel tanto experimental como clínico5,23,25,27.
Reanimación posnatal con bajo oxígenoLa utilización de concentraciones bajas de oxígeno durante la reanimación disminuye el estrés oxidativo durante la reoxigenación, la mortalidad y la tendencia a la EHI28. El uso del aire ambiente en la reanimación del recién nacido a término ha sido recomendado por la reciente edición del International Liaison Committee on Resuscitation 201529.
Alopurinol o bloqueo de la xantina oxidasaEl alopurinol es un inhibidor selectivo de la XO, de la generación de NO a partir de nitritos, un quelante de hierro libre así como un neutralizador de radicales hidroxilo. Todo ello lo convierte en un candidato apropiado para la neuroprotección21,22.
En 199830 se realizó un estudio piloto en el que se administraron 40mg/kg de alopurinol a recién nacidos con asfixia. Se observó una reducción del estrés oxidativo, una mejoría en la perfusión cerebral, de la actividad cortical cerebral, y ausencia de signos de toxicidad derivados del alopurinol. Sin embargo, en un estudio posterior31 la administración de alopurinol dentro de las 4 primeras horas después del nacimiento no mostró ningún efecto beneficioso en morbimortalidad frente a placebo, lo cual se atribuyó al retraso posnatal de su administración. Los mismos investigadores administraron alopurinol a mujeres gestantes que presentaron signos de hipoxia fetal32. Los neonatos, que alcanzaron niveles terapéuticos de alopurinol en sangre de cordón, tuvieron niveles significativamente más bajos de proteína S-100B, un marcador de daño cerebral, así como de hierro libre no unido a proteínas, un marcador de estrés oxidativo32. En estudios de seguimiento neonatal el subgrupo de recién nacidos moderadamente asfixiados que habían recibido alopurinol en las primeras horas después del parto (<4h) y una segunda dosis a las 12h tuvieron mejor resultado en la escala Wechsler Intelligence Scale for Children (WISC) que los que habían recibido placebo33. Recientemente, estos mismos autores han publicado un estudio multicéntrico y aleatorizado alopurinol/placebo administrado a gestantes con sospecha de hipoxia fetal durante el período previo al expulsivo y no pudieron confirmar una reducción significativa de S100B; sin embargo, el análisis post hoc demostró un beneficio potencial en los recién nacidos de madres tratadas con alopurinol34.
Actualmente se está realizando un estudio multicéntrico europeo (acrónimo ALBINO trial; EudraCT-2016-000222-19), aleatorizado, placebo/alopurinol en faseiii cuyo objetivo primario es la reducción de muerte y/o discapacidad y en el que participamos 18 hospitales de la red de asistencia pública de España. Se administrará alopurinol a recién nacidos con sospecha de EHI secundaria a asfixia perinatal en los primeros 30min de vida, y los que se incluyan en hipotermia recibirán una segunda dosis a las 12h después de la primera. Se incluyen estudios de metabolómica, neurofisiología e imagen, además del seguimiento neonatal hasta la edad de 24meses.
MelatoninaLa melatonina (N-acetil-5-metoxitriptamina) es una indolamina endógena que ha mostrado resultados prometedores en la EHI por sus propiedades antioxidantes, antiinflamatorias y antiapoptóticas. La acción antiinflamatoria se ejerce regulando a la baja (down-regulation) señalizadores de inflamación y la liberación del NO neuronal. La melatonina también bloquea la liberación de proteínas pro-apoptogénicas por las mitocondrias y modula la acción de GABA y receptores de glutamato35. La melatonina utilizada como coadyuvante de la hipotermia en pacientes con EHI redujo el estrés oxidativo y mejoró la supervivencia y el neurodesarrollo36–38, aunque los resultados estuvieron limitados por el tamaño muestral. Se precisan estudios con un poder estadístico adecuado para demostrar la efectividad de la melatonina y responder a preguntas pendientes.
ConclusionesLa EHI continúa siendo un grave problema en neonatología. A pesar de la generalización de la hipotermia, la morbimortalidad sigue siendo muy elevada. Las especies reactivas de oxígeno son agentes patogénicos importantes en la asfixia que evoluciona a EHI. Por lo tanto, la disminución de su producción, su neutralización o el bloqueo de las vías inflamatorias o pro-apoptóticas que activan constituyen estrategias que, combinadas con otras cuyas dianas, como son la excitotoxicidad, pro-angiogénesis o pro-neurogénesis, pueden mejorar el pronóstico de supervivencia íntegra de los neonatos con EHI.
FinanciaciónEl presente estudio ha sido financiado con la ayuda EC11-244 (ISCIII; Ministerio de Economía y Competitividad; Reino de España) concedido a MV y por un contrato a AN de la RETICS Red de Salud Materno Infantil y del Desarrollo (SAMID) financiada por el PN I+D+I 2008-2011, del ISCIII - Subdirección General de Evaluación y Fomento de la Investigación y fondos FEDER. Referencia RD16/0022/0001.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Relación de los hospitales y co-investigadores participantes en el ensayo clínico EC11-244 (Instituto Carlos III; Ministerio de Economía, Industria y Competitividad; Reino de España), con registro EudraCT-2011-005696-17
Hospital Universitario y Politécnico La Fe, Valencia: Antonio Nuñez-Ramiro; Anna Parra-Llorca; Ana García-Robles; Ana García-Blanco; María Cernada; Raquel Escrig-Fernández; Ana Gimeno-Navarro; Nuria Boronat González.
Hospital Universitario Puerta del Mar, Cádiz: Isabel Benavente; Simón Lubián López.
Hospital Regional Universitario Materno Infantil Carlos Haya, Málaga: Mercedes Chaffanel; Enrique Salguero; María del Mar Serrano Martín; Rafael Maese Heredia.
Hospital Universitario Gregorio Marañón, Madrid: Dorotea Blanco; María Arriaga.
Hospital Universitario La Paz, Madrid: Eva Valverde; Malaika Cordeiro; Adelina Pellicer; Laura Sánchez; Paloma López Ortego; Maricarmen Bravo.
Hospital Universitario 12 de Octubre, Madrid: María Teresa Moral-Pumarega; María del Carmen Pérez-Grande; Noelia Ureta Velasco; Rocío Mosqueda Peña; Ana Melgar Bonis.
Hospital Universitario de Cruces, Barakaldo: Begoña Loureiro; Jon López de Heredia; María Jesús Martinez; Cristina Iniesta.
Hospital Universitario Vall d’Hebron, Barcelona: Héctor Boix; Yolanda Castilla; Cristina Fernández-García.
Complejo Hospitalario Universitario Vigo, Vigo: Juan Fernández-Lorenzo; Eva González-Colmenero; María Luisa González-Durán; Ana Concheiro-Guisán.
Hospital Universitario Central de Asturias: Belén Fernández Colomer; Rosa Patricia Arias Llorente.
Hospital Universitario Virgen del Rocío, Sevilla: Antonio Pavón; José Fernando Ferreira López.
Hospital Universitario Reina Sofía, Córdoba: Inés Tofé; Pilar Jaraba.
Hospital Universitario Quironsalud, Madrid: Fernando Cabañas; M. Angeles Caballero; Manuela López-Azorín.

