

El síndrome de Kabuki (OMIM: #147920) presenta fisuras palpebrales largas con eversión del párpado inferior, puente nasal deprimido, cejas arqueadas, orejas displásicas y, en la mayoría de los casos, retraso mental. Los pacientes presentan anomalías menores y mayores en diferentes sistemas. Su base genética es heterogénea; recientemente se ha asociado a mutaciones del gen MLL2.
Descripción de casosSe presentan dos pacientes con características clínicas compatibles con el síndrome, siendo las principales: fisuras palpebrales largas con eversión del párpado inferior, cejas arqueadas, puente nasal deprimido, punta nasal plana, persistencia de almohadillas en pulpejos de dedos, cardiopatía y anomalías renales.
ComentariosEl diagnóstico en esta entidad es clínico; las características descritas de los casos son comparadas con los pacientes descritos en la literatura. La importancia de un diagnóstico temprano radica en el manejo preventivo y en ofrecer a la familia un asesoramiento genético adecuado.
Kabuki syndrome (OMIM: #147 920) presents as large palpebral fissures with eversion of the lateral third of the lower eyelids, depressed nasal bridge, arched eyebrows, dysplastic ears and in most cases, with mental retardation. Patients have minor and major abnormalities in different systems. Its genetic basis is heterogeneous, but recently has been associated with mutations in gen MLL2.
Case reportsWe present two patients with clinical features compatibles with the syndrome, mainly: large palpebral fissures with eversion of the lateral third of the lower eyelids, depressed nasal bridge, arched eyebrows, flat nose, persistent fingertip pads, cardiopathies and renal anomalies.
CommentaryThe diagnosis of this condition is clinical. The characteristics in the cases are compared with the patients reported in the literature. The importance of early diagnosis is to provide preventive management and an appropriate genetic counseling for the family.
El síndrome de Kabuki (KMS [OMIM #147920]), o síndrome Niikawa-Kuroki, es una patología con múltiples anomalías asociadas a retraso mental. Descrita inicialmente de forma independiente en 1981 por los doctores Niikawa1 y Kuroki2, fue nombrado por la semejanza facial de los individuos afectados con el maquillaje de los actores de Kabuki (teatro tradicional japonés)1. Actualmente, la palabra maquillaje no es usada por considerarse un término despectivo3.
En 1988, Niikawa et al.4 describieron 62 casos y establecieron las bases del diagnóstico de KMS, según 5 principales manifestaciones: características faciales, anomalías esqueléticas, alteración en los dermatoglifos, retraso mental y talla baja. Sobre esta base, Matsutmoto et al.5 categorizaron estos rasgos como mayores y menores para facilitar su identificación. Dentro de los rasgos mayores se encuentran: fisuras palpebrales largas con eversión del párpado inferior, puente nasal deprimido, cejas arqueadas, almohadillas en el pulpejo de los dedos, pabellones auriculares prominentes o malformadas, acortamiento del quinto dedo, paladar alto o hendido, dentadura anormal, hipotonía, retraso mental, talla baja, ptosis e hipoacusia. Los rasgos menores se caracterizan por: escleras azules, escoliosis, anomalías cardiovasculares, malformaciones renales, vértebras deformadas, criptorquidia y deficiencia de la hormona del crecimiento, entre otros.
Se ha estimado una prevalencia del KMS entre 1/86.0006 y 1/32.0004,7,8. Se han descrito más de 400 casos en todo el mundo3,5,9,10; en América Latina, los casos publicados han aumentado considerablemente en los últimos años11,12. Se presenta en ambos sexos y todas las razas. La mayoría de los casos son de presentación esporádica, aunque se han descrito algunos con transmisión autosómica dominante. Su base genética es heterogénea; el estudio citogenético convencional suele ser normal en la mayoría de los casos. Las pruebas moleculares han identificado mutaciones en el gen TRPM3, en 9q21.11/9q21.12, en casos con fisura palatina13. Alteraciones tipo duplicaciones en 8p22/8p23.1 encontradas en algunos pacientes14,15 no han sido descritas posteriormente8,16,17. Recientemente, se han referido mutaciones del gen MLL2 como una de las principales causas en pacientes con el síndrome; este estudio se ha llevado a cabo mediante una técnica novedosa aplicada en clínica: el análisis del exoma18.
Se describen los hallazgos de 2 casos clínicos comparándolos con la literatura, para ayudar a la oportuna identificación del KMS y ofrecer un manejo adecuado.
Caso clínico 1Paciente de 7 años y 9 meses, procedente de Chipatá (Departamento de Santander, Colombia), producto de primera gestación, nacido por cesárea por sufrimiento fetal agudo, hospitalizado por hipoglucemia, trombocitopenia transitoria e ictericia. Hallaron las siguientes malformaciones: coartación de aorta, agenesia renal derecha y paladar hendido. Fue intervenido quirúrgicamente en varias oportunidades. Presentó otitis media crónica. El desarrollo psicomotor fue normal; actualmente cursa segundo grado de primaria con buen rendimiento escolar. No hay antecedentes familiares patológicos ni consanguinidad. Las medidas antropométricas fueron: peso 15 kg (<percentil 5), talla 104cm (<percentil 5), perímetro cefálico (PC) 49cm (percentil 3-10) corregido para la talla, distancia interpupilar (DIP) 5cm (percentil 25), distancia intercantal interna (DICI) 2,4cm (percentil 3), distancia intercantal externa (DICE) 8cm (percentil 25-50), mano total (MT) 11cm (<percentil 3), dedo medio (DM) 4,5cm (<percentil 3), DM/MT 0,409 (percentil 3-25) y fisura palpebral 3,2cm (>percentil 95).
Los hallazgos positivos encontrados fueron:
- 1.
Talla baja para la edad.
- 2.
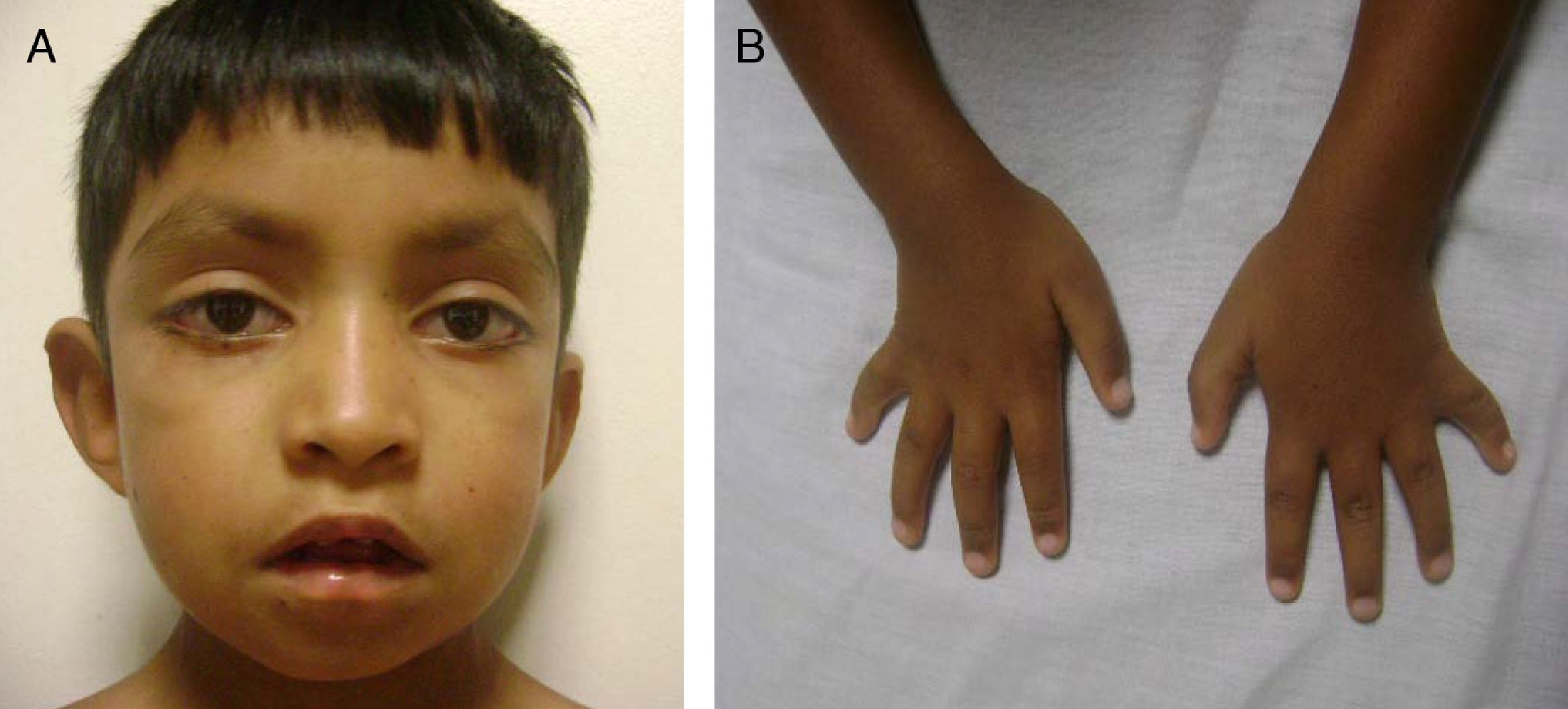
Cráneo-facial: cabello con implantación baja, hipertricosis, cejas arqueadas y tercio externo de las cejas ralo, fisuras palpebrales largas con eversión del párpado inferior, punta nasal plana, paladar alto y cicatriz de corrección quirúrgica, pabellones auriculares normoimplantados en copa (fig. 1A).
- 3.
Tórax y abdomen: tórax simétrico con pectus carinatum en el tercio inferior, exploración cardiopulmonar sin alteraciones; hernia periumbilical.
- 4.
Extremidades: braquidactilia, clinodactilia del quinto dedo de ambas manos, persistencia de las almohadillas del pulpejo de los dedos (fig. 1B).
 Se observan las cejas arqueadas y con el tercio externo disperso. Puente nasal deprimido. Fisuras palpebrales largas, con eversión del párpado inferior. B) Se observan la braquidactilia y la clinodactilia del quinto dedo de ambas manos.")
Se realiza cariotipo por técnica de bandas GTG de 550 bandas con el resultado: 46,XY. Aumento del tamaño de la región heterocromatina del brazo largo del cromosoma 1 (1qh+).
Caso clínico 2Paciente de 8 años y 11 meses, procedente de Floridablanca (Departamento de Santander, Colombia). Producto de segunda gestación. Nacido por cesárea por sufrimiento fetal agudo, presentó cianosis e hipoxia perinatal, requiriendo conexión a ventilación mecánica; además, presentó ictericia, permaneciendo 26 días hospitalizado. Se hallaron las siguientes malformaciones: hipoplasia renal derecha, comunicación interventricular y aorta bicúspide con insuficiencia leve. Se identifican dismorfismo facial y retraso del desarrollo psicomotor. Presentó otitis a repetición y bronquitis en 6 oportunidades. Ha recibido hormona de crecimiento. No hay antecedentes familiares patológicos ni consanguinidad. Las medidas antropométricas fueron: peso: 30 kg (percentil 50-75), talla 121cm (<percentil 5), PC: 50cm (percentil 3-10) corregido para la talla, DIP: 3cm (percentil 3-25), DICI: 2,6cm (percentil 25), DICE: 8,2cm (percentil 75), MT: 13,6cm (percentil 25), DM: 5,2cm (<percentil 3), DM/MT: 0,38 (<percentil 3) y fisura palpebral 2,7cm (percentil 75).
Los hallazgos positivos fueron:
- 1.
Talla baja proporcionada.
- 2.
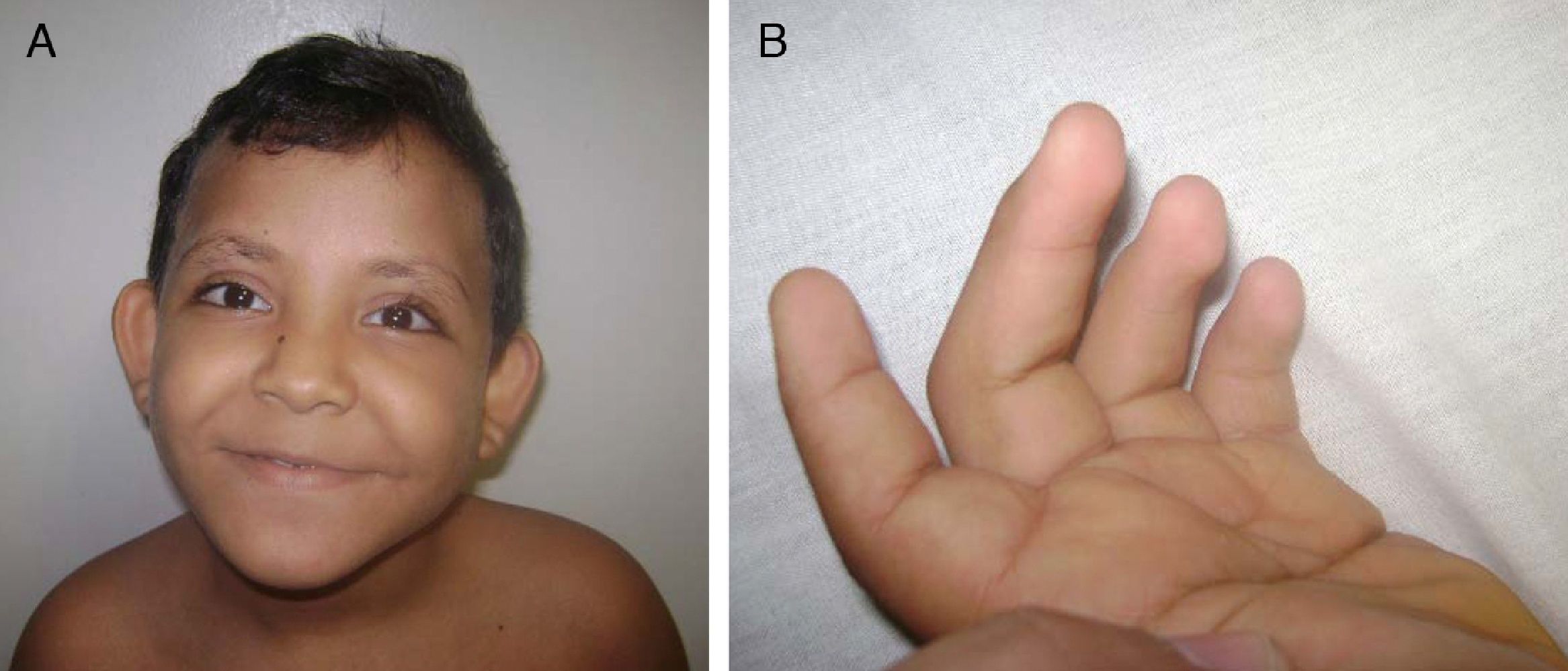
Cráneo-facial: fisuras palpebrales largas con eversión del párpado inferior, cejas con tercio externo ralo, pabellones auriculares alados de baja implantación, punta nasal plana, comisura labiales dirigidas hacia abajo, mal oclusión y diastema en piezas dentales, y cuello móvil sin lesiones. (fig. 2A)
- 3.
Extremidades: simétricas, braquidactilia, persistencia de almohadilla del pulpejo de los dedos, pliegue palmar único bilateral y clinodactilia del quinto dedo (fig. 2B).
 Se observan las cejas arqueadas y con el tercio externo disperso. Puente nasal deprimido. Fisuras palpebrales largas, con eversión del párpado inferior, orejas en copa. B). Se observa la persistencia de la almohadilla en los dedos de la mano.")
Se realiza cariotipo por técnica de bandas GTG de 700 bandas con resultado: 46,XY, sin alteraciones numéricas ni estructurales.
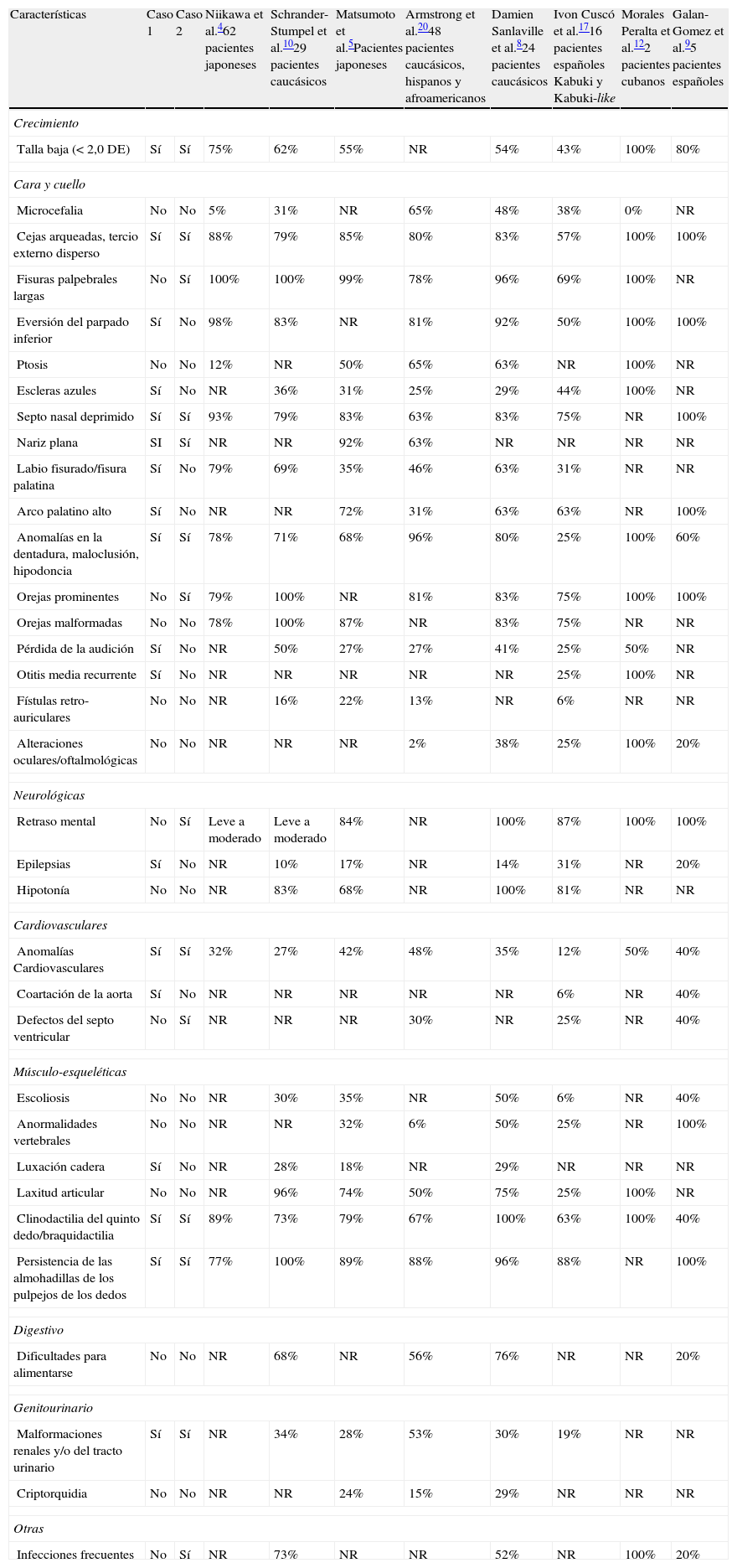
DiscusiónEl síndrome de Kabuki presenta anomalías faciales que usualmente lo definen1,2,7,10. En la tabla 1 se presentan las alteraciones descritas en la literatura comparándolas con los pacientes de este artículo. En cuanto al perímetro cefálico, lo común es la presencia de microcefalia; sin embargo, puede presentarse normocefalia con valores bajos, como lo sucedido con los casos presentados. El retraso mental ha sido descrito en varios pacientes; sin embargo, no es condicionante para establecer o excluir el diagnóstico. En la tabla 1 se puede apreciar que algunos artículos refieren esta condición en el 100% de los pacientes y en otros se da un poco más del 80%. Uno de los pacientes descritos en este artículo tiene los criterios clínicos para el síndrome pero con desarrollo neurológico normal. Existen signos con frecuencia alta que deben tenerse en consideración al evaluarse a un paciente con esta patología como son: talla baja, cejas arqueadas con tercio externo disperso, fisuras palpebrales largas, eversión del párpado inferior, septo nasal deprimido, nariz plana, anormalidades en la dentadura, maloclusión, hipodoncia, orejas prominentes, clinodactilia y persistencia de almohadilla en los pulpejos de los dedos; la mayoría de estos están presentes en los pacientes descritos en este artículo. Por otro lado, hay anomalías en diferentes sistemas que se presentan con menor frecuencia pero que deben descartarse en todo paciente con diagnóstico clínico de síndrome de Kabuki, dentro de las cuales están: malformaciones del sistema cardiovascular y genitourinario como lo refieren varios de los casos previamente publicados10,17,19,20.
Hallazgos clínicos de nuestros pacientes comparados con los datos de la literatura
| Características | Caso 1 | Caso 2 | Niikawa et al.462 pacientes japoneses | Schrander-Stumpel et al.1029 pacientes caucásicos | Matsumoto et al.5Pacientes japoneses | Armstrong et al.2048 pacientes caucásicos, hispanos y afroamericanos | Damien Sanlaville et al.824 pacientes caucásicos | Ivon Cuscó et al.1716 pacientes españoles Kabuki y Kabuki-like | Morales Peralta et al.122 pacientes cubanos | Galan-Gomez et al.95 pacientes españoles |
| Crecimiento | ||||||||||
| Talla baja (<2,0 DE) | Sí | Sí | 75% | 62% | 55% | NR | 54% | 43% | 100% | 80% |
| Cara y cuello | ||||||||||
| Microcefalia | No | No | 5% | 31% | NR | 65% | 48% | 38% | 0% | NR |
| Cejas arqueadas, tercio externo disperso | Sí | Sí | 88% | 79% | 85% | 80% | 83% | 57% | 100% | 100% |
| Fisuras palpebrales largas | No | Sí | 100% | 100% | 99% | 78% | 96% | 69% | 100% | NR |
| Eversión del parpado inferior | Sí | No | 98% | 83% | NR | 81% | 92% | 50% | 100% | 100% |
| Ptosis | No | No | 12% | NR | 50% | 65% | 63% | NR | 100% | NR |
| Escleras azules | Sí | No | NR | 36% | 31% | 25% | 29% | 44% | 100% | NR |
| Septo nasal deprimido | Sí | Sí | 93% | 79% | 83% | 63% | 83% | 75% | NR | 100% |
| Nariz plana | SI | Sí | NR | NR | 92% | 63% | NR | NR | NR | NR |
| Labio fisurado/fisura palatina | Sí | No | 79% | 69% | 35% | 46% | 63% | 31% | NR | NR |
| Arco palatino alto | Sí | No | NR | NR | 72% | 31% | 63% | 63% | NR | 100% |
| Anomalías en la dentadura, maloclusión, hipodoncia | Sí | Sí | 78% | 71% | 68% | 96% | 80% | 25% | 100% | 60% |
| Orejas prominentes | No | Sí | 79% | 100% | NR | 81% | 83% | 75% | 100% | 100% |
| Orejas malformadas | No | No | 78% | 100% | 87% | NR | 83% | 75% | NR | NR |
| Pérdida de la audición | Sí | No | NR | 50% | 27% | 27% | 41% | 25% | 50% | NR |
| Otitis media recurrente | Sí | No | NR | NR | NR | NR | NR | 25% | 100% | NR |
| Fístulas retro-auriculares | No | No | NR | 16% | 22% | 13% | NR | 6% | NR | NR |
| Alteraciones oculares/oftalmológicas | No | No | NR | NR | NR | 2% | 38% | 25% | 100% | 20% |
| Neurológicas | ||||||||||
| Retraso mental | No | Sí | Leve a moderado | Leve a moderado | 84% | NR | 100% | 87% | 100% | 100% |
| Epilepsias | Sí | No | NR | 10% | 17% | NR | 14% | 31% | NR | 20% |
| Hipotonía | No | No | NR | 83% | 68% | NR | 100% | 81% | NR | NR |
| Cardiovasculares | ||||||||||
| Anomalías Cardiovasculares | Sí | Sí | 32% | 27% | 42% | 48% | 35% | 12% | 50% | 40% |
| Coartación de la aorta | Sí | No | NR | NR | NR | NR | NR | 6% | NR | 40% |
| Defectos del septo ventricular | No | Sí | NR | NR | NR | 30% | NR | 25% | NR | 40% |
| Músculo-esqueléticas | ||||||||||
| Escoliosis | No | No | NR | 30% | 35% | NR | 50% | 6% | NR | 40% |
| Anormalidades vertebrales | No | No | NR | NR | 32% | 6% | 50% | 25% | NR | 100% |
| Luxación cadera | Sí | No | NR | 28% | 18% | NR | 29% | NR | NR | NR |
| Laxitud articular | No | No | NR | 96% | 74% | 50% | 75% | 25% | 100% | NR |
| Clinodactilia del quinto dedo/braquidactilia | Sí | Sí | 89% | 73% | 79% | 67% | 100% | 63% | 100% | 40% |
| Persistencia de las almohadillas de los pulpejos de los dedos | Sí | Sí | 77% | 100% | 89% | 88% | 96% | 88% | NR | 100% |
| Digestivo | ||||||||||
| Dificultades para alimentarse | No | No | NR | 68% | NR | 56% | 76% | NR | NR | 20% |
| Genitourinario | ||||||||||
| Malformaciones renales y/o del tracto urinario | Sí | Sí | NR | 34% | 28% | 53% | 30% | 19% | NR | NR |
| Criptorquidia | No | No | NR | NR | 24% | 15% | 29% | NR | NR | NR |
| Otras | ||||||||||
| Infecciones frecuentes | No | Sí | NR | 73% | NR | NR | 52% | NR | 100% | 20% |
NR: no registra.
El cariotipo de bandas G de alta resolución suele ser normal en la mayoría de los casos. El cariotipo del caso clínico 1 exhibe un aumento de la región heterocromatina 1qh+, considerado una variante normal. Por su parte, en el caso clínico 2 el cariotipo fue normal, aunque mediante esta técnica no se puede descartar la presencia de pequeños reordenamientos cromosómicos o bajos niveles de mosaicismo, como lo descrito en otros pacientes con fenotipo Kabuki17. En los pacientes no se han realizado estudios de hibridación genómica comparativa (CGH), ni análisis molecular del gen MLL2 recientemente implicado, siendo el paso a seguir para poder identificar la etiología y con esto contribuir con la información para establecer la posibilidad de correlación genotipo-fenotipo18.
El consejo genético es complicado, por la heterogeneidad en su base genética, por lo que la mayoría de casos son esporádicos; sin embargo, se han descrito pacientes con mecanismos de herencia autosómica dominante. Si los padres no tienen características, se debe considerar una mutación de novo; el riesgo de recurrencia sería menor del 1%, pero el individuo afectado podría tener una probabilidad del 50% de transmitir la condición.
La guía de prevención para el síndrome ya está establecida21, basada en las posibles complicaciones en función de diferentes edades. En la infancia temprana es importante realizar evaluaciones que permitan descartar o confirmar la presencia de malformaciones en órganos internos, principalmente el corazón, la arteria aorta y el sistema genitourinario. Además debe realizarse la prevención y/o rehabilitación de otros sistemas afectados como son el neurológico, fonoaudiológico, pulmonar, osteomuscular y endocrinológico. Por este motivo, el abordaje es interdisciplinario y el objetivo es ofrecer un mejor pronóstico y calidad de vida.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

