


Sr. Editor:
La alfa-manosidosis es una enfermedad de depósito infrecuente, resultado del déficit de la enzima alfa-manosidasa lisosomal. Este defecto produce una acumulación de manosilglucoproteínas, fundamentalmente en el sistema nervioso central y en el hígado, junto con una excreción aumentada de manosiloligosacáridos en orina1,2. Estos pacientes habitualmente presentan fenotipo hurleriano, retraso del desarrollo psicomotor, disóstosis múltiples y hepatoesplenomegalia, y es menos frecuente la cifoescoliosis, craneosinostosis, defectos de audición, opacidades corneales y hernias de la pared abdominal3–5.
El diagnóstico en una primera aproximación consiste en detectar la presencia de manosiloligosacáridos en la orina, aunque el diagnóstico definitivo se establece demostrando la deficiencia de alfa-manosidasa.
Varón de 4 años valorado en nuestra unidad por retraso del lenguaje de tipo mixto, con predominio del déficit expresivo y mala pronunciación. Tenía, además, moderado retraso psicomotor.
Antecedentes familiares y personales: padres no consanguíneos. Dos hermanos sanos. Embarazo controlado y tolerado. Parto a término. Somatometría normal y retraso en adquisición de hitos motores y cognitivos.
Exploración física: peso, 22kg (P90); talla, 109cm (P75); perímetro craneal, 53cm (P97).
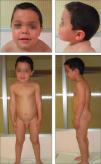
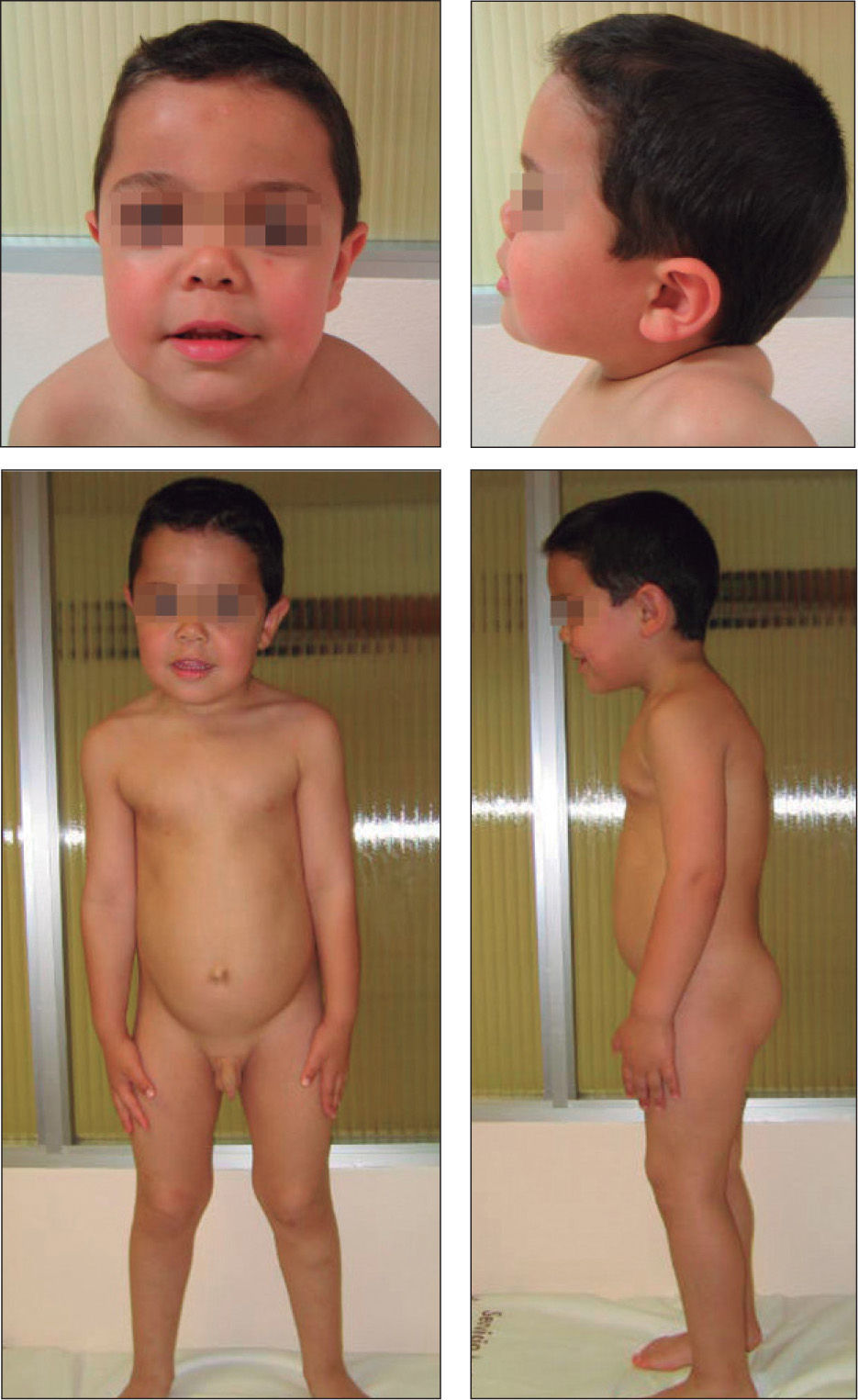
No presentaba alteraciones dermatológicas. Sus rasgos faciales eran toscos, con raíz nasal deprimida, hipertelorismo, pabellones auriculares de implantación baja y posteriores, frente amplia y aspecto macrocéfalo (fig. 1). La auscultación cardíaca y pulmonar fueron normales. Presentaba hepatomegalia de 2cm bajo reborde costal derecho y hernia umbilical. El resto de la exploración no mostró hallazgos patológicos. El examen oftalmológico fue normal.
Exámenes complementarios: Hemograma y bioquímica sanguínea: sin hallazgos patológicos. Potenciales evocados auditivos: hipoacusia neurosensorial con onda v a una intensidad de estimulación de 50dB. Estudios de imagen: la ecografía abdominal mostró hepatoesplenomegalia con parénquima normal. La serie ósea no tuvo hallazgos de interés, aunque sí retraso de edad ósea, que era de 3 años y 6 meses. Evaluación psicopedagógica: el inventario de desarrollo de Battelle mostró un moderado retraso global del desarrollo, especialmente en el área cognitiva con una edad mental equivalente a unos 28 meses aproximadamente (más de 2 años por debajo de la cronológica). En cuanto al área motora, el retraso fue leve. Estudios metabólicos: los glucosaminoglicanos en la orina fueron normales. Los oligosacaridos en la orina obtenidos mediante cromatografía en capa fina indicaron aumento de la excreción de algunos oligosacáridos enriquecidos en residuos de manosa, con un patrón cromatográfico típico de los pacientes afectados de esta enfermedad. El estudio enzimático en suero mostró un déficit de alfa-manosidasa: 0,04 nmoles/min/ml (5 % respecto al control). Estos resultados confirmaron el diagnóstico de alfa-manosidosis.
Las enfermedades lisosomales son un amplio grupo de errores congénitos del metabolismo originados por la deficiencia de una enzima lisosomal, infrecuentes de manera aislada, que muestran una amplia heterogeneidad clínica. Se clasifican según el sustrato acumulado: esfingolipidosis, mucopolisacaridosis, mucolipidosis y defectos de la degradación de las glucoproteínas. Las más frecuentes en la población española son las enfermedades de Gaucher, Hurler, Hunter y Fabry. Muchas de ellas cursan con un fenotipo dismórfico hurleriano que, cuando es evidente, permite hacer el diagnóstico con relativa facilidad mediante el estudio metabólico pertinente.
La alfa-manosidosis es una glucoproteinosis debida a un déficit de la alfa manosidasa, enzima necesaria para el catabolismo de las cadenas oligosacáridas que forman parte de las N-glucoproteínas. Es éste un trastorno infrecuente, con una prevalencia estimada de 1 caso por 500.0006.
Clásicamente se describen dos tipos de alfa-manosidosis3. El tipo I, o forma infantil, es un cuadro grave, de inicio entre los 3 y 12 meses de vida. Se caracteriza por retraso mental y motriz graves, rasgos toscos, hepatoesplenomegalia, disóstosis múltiple, cataratas, opacidades corneales y sordera, y cursa con deterioro progresivo y muerte precoz. El tipo II, o forma juvenil, se inicia entre el primer y cuarto año de vida con retraso mental y pérdida auditiva progresiva. El cuadro tiene un curso prolongado y se extiende hasta la vida adulta, con posibilidad de evolucionar a hidrocefalia (como en las mucopolisacaridosis) y paraplejia espástica.
Sin embargo, hoy en día se admite que hay un continuum entre estos dos tipos y, por lo tanto, la clínica puede ser muy variable. Así, aunque los síntomas puedan ser poco expresivos en la cara y el esqueleto, en la primera consulta -momento en el que se suele solicitar atención por trastornos del lenguaje, dificultades cognitivas, trastornos del aprendizaje o problemas de conducta- debemos guiarnos por la exploración detallada de rasgos ligeramente toscos, lo cual nos pondrán en alerta sobre la posibilidad de estar ante alguna de estas enfermedades.
Esto es lo que ocurrió con nuestro paciente, que presentaba rasgos faciales levemente toscos y un discreto fenotipo de mucopolisacaridosis. Estos factores, junto con un moderado retraso del lenguaje, esplenomegalia y hernia umbilical, sugirieron una posible enfermedad de depósito lisosomal. La oligosacariduria y la reducción de actividad de la alfa-manosidasa condujeron al diagnóstico de alfa-manosidosis.
El diagnóstico no requirió procedimientos invasivos7 y el suero y el plasma fueron útiles para demostrar un déficit grave de la enzima (0 %-15 %). El déficit parcial de alfa-manosidasa se pudo confirmar también en leucocitos y/o en cultivo fibroblastos. El patrón hereditario es autosómico recesivo, y se localizó el defecto génico en el cromosoma 19 (19 p13.2-q12)3. Estudios realizados por Sbaragli et al8 han demostrado la existencia de nuevas mutaciones del gen MAN2B1 responsables del déficit enzimático, que ponen de manifiesto un alto grado de heterogeneidad mutacional en la alfa-manosidosis, comparable al observado en muchos otros trastornos lisosomales.
En la actualidad no existe, a diferencia de otras enfermedades lisosomales, como la enfermedad de Gaucher tipo I o de Fabry, un tratamiento efectivo. Existen varios ensayos terapéuticos entre los que destaca el trasplante de células madre hematopoyéticas con resultados hasta ahora poco alentadores, mientras que la reposición enzimática sólo es, por el momento, una hipótesis. El diagnóstico prenatal es posible mediante la biopsia de vellosidades coriales o por cultivo de amniocitos9.
En conclusión, queremos destacar que, en niños con retrasos del lenguaje y rasgos faciales toscos, es preciso realizar en la valoración diagnóstica el estudio de oligosacáridos en orina -además del de los mucopolisacáridos-, lo cual nos puede permitir, como en nuestro caso, diagnosticar una enfermedad lisosomal, como la alfa-manosidosis10.
