





La enfermedad mieloproliferativa transitoria neonatal y la leucemia aguda megacarioblástica del síndrome de Down se consideran manifestaciones distintas de la misma enfermedad. La mayoría de casos de enfermedad mieloproliferativa transitoria no requiere tratamiento mientras que la leucemia aguda megacarioblástica del síndrome de Down se caracteriza por una elevada sensibilidad a la quimioterapia, lo que ha llevado a la reducción en la intensidad de dosis de tratamiento administrada. Ambas entidades comparten mutaciones específicas en los exones 2 y 3,1 del factor de transcripción GATA1.
Pacientes y métodosHemos analizado los hallazgos biológicos incluyendo la presencia de mutaciones de GATA1 en cuatro pacientes con enfermedad mieloproliferativa transitoria neonatal (2) y leucemia aguda megacarioblástica (2) incluyendo un paciente fenotípicamente normal portador de un mosaicismo para la trisomía 21.
ResultadosEn los cuatro casos hemos encontrado la presencia de una clona GATA1 mutante y en tres de ellos se describe una mutación puntual en el exón 2 de dicho gen. Dada la heterogeneidad fenotípica de los blastos megacariocíticos y el bajo porcentaje de estos elementos, la detección de mutaciones en GATA1 resultó de gran utilidad para establecer el diagnóstico. Además, sucesivos resultados normales del análisis mutacional de GATA1 permitieron establecer la remisión molecular en 2 pacientes.
ConclusionesConcluimos que el análisis mutacional de GATA1 es una herramienta útil para el diagnóstico y manejo de los trastornos mieloproliferativos asociados a la trisomía 21.
Neonatal transient myeloproliferative disorder and acute megakaryoblastic leukaemia of Down syndrome are considered different manifestations of the same disease. In most cases, transient myeloproliferative disorders require no treatment, while acute megakaryoblastic leukaemia of Down's syndrome is characterised by an increased sensitivity to chemotherapy and its treatment should be adapted with a reduction in dose intensity. Both entities share specific mutations at exón 2 of the transcription factor GATA1.
Patients and methodsWe analysed biological features and GATA1 mutations in 4 patients with transient abnormal myelopoiesis (2) and acute megakaryoblastic leukaemia (2) including one phenotypically normal trisomy 21 mosaicism. We found abnormal GATA1 mutated clones in each case, and a specific point mutation at exón 2 was detected in three cases. Given the heterogeneous phenotype of megakaryoblastic blasts and the low percentage of blasts at presentation, the recognition of GATA1 mutations was helpful for diagnosis. In addition, molecular remission was established in 2 patients after subsequent normal mutational GATA1 analysis.
ConclusionsWe conclude that GATA1 mutational study is a useful tool for the diagnosis and management of trisomy 21 associated myeloproliferative disorders.
La trisomía 21 predispone a la leucemia y los niños con síndrome de Down (SD) tienen un riesgo 10–20 veces superior al resto de la población de desarrollar leucemia1–3. La incidencia de leucemia aguda linfoblástica en estos pacientes es 20 veces superior a la del resto de la población. Sin embargo, el incremento de incidencia más notable ocurre con la leucemia aguda megacarioblástica (LAMK) en niños de corta edad con SD, con un riesgo 500 veces superior que el resto de la población pediátrica1,2,4. La LAMK del SD muestra una tasa de respuesta al tratamiento superior al resto de la población5–9, lo que se corresponde con un marcado incremento de sensibilidad in vitro de los blastos leucémicos a diferentes agentes quimioterápicos, en particular a arabinósido de citosina6,7,10. Sin embargo, los niños con SD mayores de 5 años con leucemia aguda mieloblástica, incluyendo la LAMK, tienen un pronóstico similar al resto de pacientes7,8,11,12.
Por otro lado, los niños con SD muestran mayor sensibilidad a la toxicidad derivada de la quimioterapia y una tasa superior de mortalidad precoz durante el tratamiento7,8,13. Ambos hechos han conducido a modificar los esquemas de tratamiento de la LAMK en este grupo particular de niños menores de cinco años con SD, excluyéndoles de la indicación de transplante hematopoyético y adaptando la intensidad de la quimioterapia5–7,11,13–16.
En pacientes con SD, esta enfermedad puede venir precedida por un síndrome preleucémico transitorio en el periodo neonatal que habitualmente se resuelve espontáneamente, conocido como leucemia transitoria, mielopoiesis anormal transitoria o enfermedad mieloproliferativa transitoria neonatal (EMTN)2,6.
Además, en la mayoría de niños con SD, la LAMK viene precedida por un trastorno de la diferenciación megacarioblástica conocido como síndrome mielodisplásico (SMD) durante el que se reduce el número de plaquetas circulantes mientras aumenta el número de megariocitos de morfología anormal en la médula ósea. Este proceso tiene una duración variable que puede ser hasta de meses pero de forma inevitable desembocará en una LAMK2,3,5–7,17
Recientemente se ha comprobado que los blastos leucémicos tanto en la EMTN como en el SMD y la LAMK de los niños con SD son portadores de mutaciones adquiridas específicas del gen GATA1 ubicado en el cromosoma X que codifica el factor de transcripción hematopoyético GATA11–3,6,7,18,19. Ello hace que el análisis mutacional de GATA1 resulte de gran utilidad al permitir establecer con fidelidad el diagnóstico de trastorno mieloproliferativo asociado a SD1,20, facilitando la orientación terapéutica: actitud expectante en los casos de EMTN y tratamiento «adaptado» en los casos de LAMK.
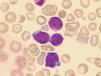
Pacientes y métodosHemos investigado la presencia de mutaciones en los exones 2 y 3,1 de GATA1 en dos pacientes diagnosticados de EMTN y otros dos diagnosticados de LAMK (tabla 1). Los casos de EMTN se diagnosticaron en su primer día de vida como consecuencia de una elevación del recuento de leucocitos (98 y 39,8×109/l) y la identificación de blastos mieloides en sangre periférica (36,2 y 9,9×109/l) (tabla 1, fig. 1) en dos pacientes con fenotipo Down en los que posteriormente se confirmó la trisomía 21. En uno de ellos no se encontraron citopenias al debut mientras el otro mostraba trombocitopenia con un recuento de plaquetas de 22×109/l. Ambos eran recién nacidos pretérmino de 33 y 36 semanas de gestación respectivamente, requiriendo el primero de ellos cuidados intensivos neonatales por insuficiencia respiratoria y un cuadro de hidrops. Los 2 diagnósticos de LAMK se establecieron en dos niñas de 19 y 30meses de edad. La primera había sido diagnosticada en el periodo neonatal de un mosaicismo para la trisomía 21 y fue remitida a nuestro centro por púrpura trombocitopénica (recuento de plaquetas de 8×109/l). La segunda paciente se había diagnosticado en nuestra unidad 14meses antes de un SMD relacionado con el SD en un aspirado medular practicado por detectarse trombocitopenia y en el que se encontró hiperplasia megacariocítica con rasgos displásicos en ausencia de blastos. Las muestras de los cuatro pacientes fueron examinadas en busca de mutaciones en los exones 2 y 3,1 de GATA1 (mutaciones previas al codón 84). La extracción del ADN de las muestras de sangre periférica y/o médula ósea se realizó empleando el Mini Kit QIAamp DNA (Qiagen®) según las instrucciones del fabricante o bien mediante una técnica estándar de extracción en fenol-cloroformo. Para el análisis mutacional del exón 2 de GATA1 se empleó la técnica de reacción en cadena polimerasa (PCR) mediante cebadores previamente diseñados por el Dr. Fisher en el Instituto Weatheral de Medicina Molecular (datos no publicados) y para el exón 3,1 se emplearon los pares de cebadores previamente descritos por Wechsler et al.21 Las muestras amplificadas se analizaron mediante cromatografía líquida de alta resolución (WAVE; Transgenomic®, Omaha, NE). Los fragmentos del exón 2 se analizaron a 60,2, 61,2 y 62,2°C y los fragmentos del exón 3,1 a 61,9 y 62,9°C. La secuenciación bidireccional se realizó mediante la tecnología ABI BigDye® y se analizó empleando los programas Sequencher® y Mac Vector®. En los casos en los que el análisis de la secuenciación no resultaba claro se realizó un subclonaje de los productos de la PCR empleando el vector pGEM-T Easy (Promega®) según el protocolo del fabricante, seguido de una secuenciación de los productos clonados.
Característica clínico-biológicas y evolución de los cuatro pacientes
| Pacientes | 1 | 2 | 3 | 4 |
| Edad | 1 día | 1 día | 19 meses | 30 meses |
| Diagnóstico | EMTN | EMTN | LAMK | LAMK |
| Leucocitos (×109/l) | 98 | 39,8 | 7,9 | 4,9 |
| Mieloblastos (%)a | 37 | 25 | 38 | 48 |
| Hb | 17 | 19,8 | 8,8 | 8,7 |
| Plaquetas (×109/l) | 450 | 22 | 8 | 14 |
| Inmunofenotipo | ||||
| CD45 | + | + | + | + |
| CD34 | ++ | + | + | + |
| CD117 | + | + | + | + |
| CD11b | − | − | + | + |
| CD13 | − | − | + | + |
| CD33 | +/−b | + | + | + |
| CD64 | −/+b | − | − | − |
| CD38 | −/+b | + | + | + |
| MPO | − | − | − | − |
| HLA-DR | −/+b | − | − | + |
| CD36 | +/−b | + | + | + |
| CD41 | ND | − | ND | ND |
| CD42 | ND | + | ND | ND |
| CD61 | ++/−b | ++ | − | + |
| CD5 | ND | + | ND | − |
| CD7 | + | + | + | + |
| CD56 | + | − | + | − |
| Citogenéticac | «47,XX,+21» [21] | «47,XY,+21» [20] | «48,XX,+21» [3] | «48,XX,+8,+21» [12] |
| «46,XX» [16] | ||||
| GATA1d | Posición 147 | Posición 266 | Indetectable | Posición 161 |
| Manejoe | Conservador | Conservador | AML-BFM 98 | AML-BFM 98 |
| Evolución | Remisión espontánea | Remisión espontánea | Remisión completa | Exitus (neumonitis intersticial) |
| (60días) | (100días) | |||
| Seguimiento (meses)f | 18 | 36 | 36 | 1 |
EMTN: enfermedad mieloproliferativa transitoria neonatal; LAMK: leucemia aguda megacarioblástica; ND: no disponible
Porcentaje de mieloblastos en el examen citológico de sangre periférica para los pacientes 1 y 2 y de médula ósea para los pacientes 3 y 4.
Fórmula cromosómica y número de metafases analizadas en sangre periférica para los pacientes 1 y 2 y en médula ósea para los pacientes 3 y 4. En el paciente 3 se identifica una doble población (mosaicismo).
Sustituciones puntuales en exón 2. En el paciente 3 la cromatografía mostraba cambios sutiles sugestivos de la presencia de una pequeña clona GATA1 mutante.
Los dos pacientes diagnosticados de EMTN recibieron tratamiento de soporte incluyendo cuidados intensivos y ventilación mecánica en el paciente 1 (hidrops). Los dos pacientes diagnosticados de LAMK recibieron quimioterapia de intensidad reducida según el protocolo AML-BFM 9810.
Presentamos los hallazgos inmunofenotípicos, citogenéticos y moleculares de estos cuatro pacientes.
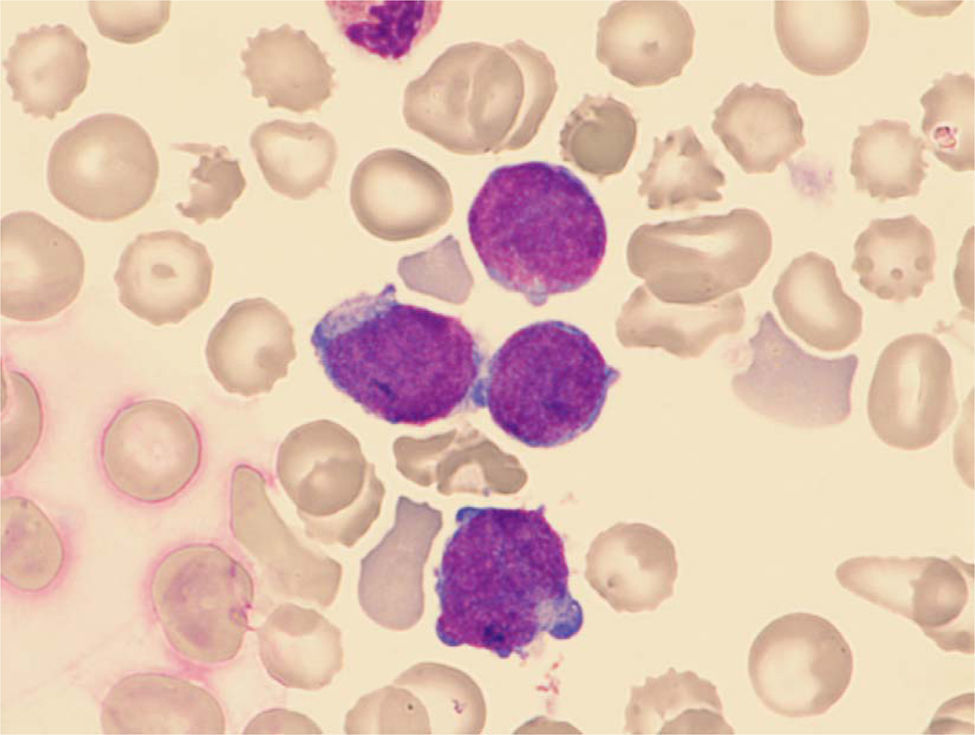
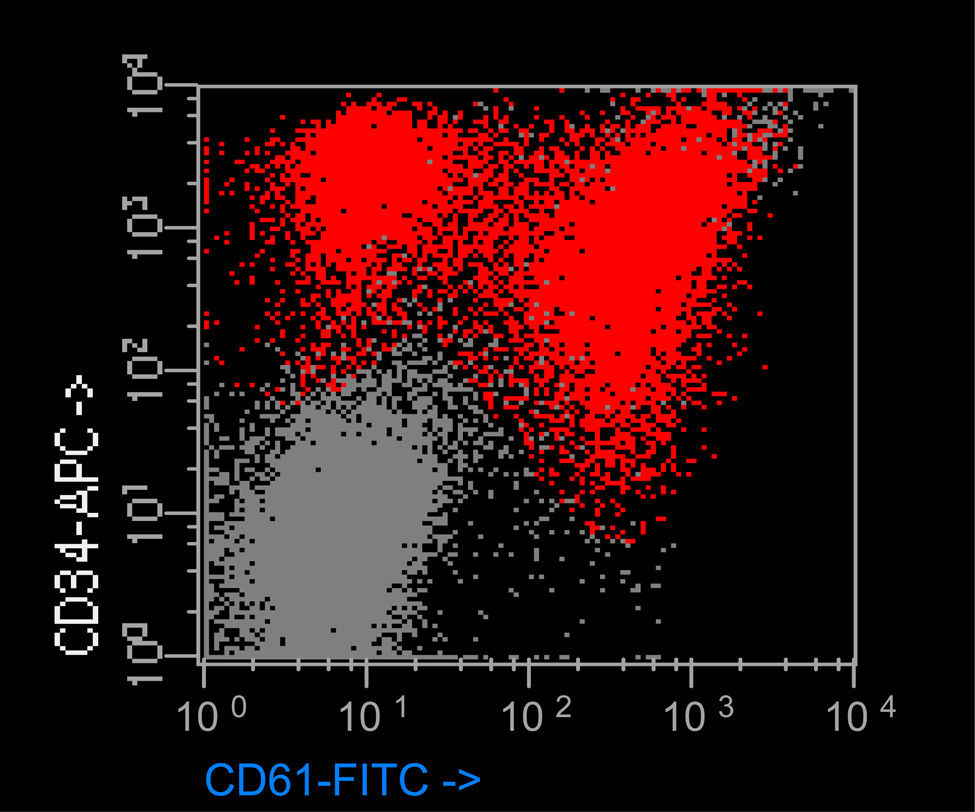
ResultadosEl porcentaje de blastos en sangre periférica en los dos pacientes con EMTN fue 37 y 25%. Un paciente mostró una doble población blástica tanto por citología óptica como por técnicas de fluorocitometría (fig. 2). En los dos pacientes diagnosticados de LAMK el porcentaje de blastos en el aspirado medular fue de 38 y 48% respectivamente en el examen citológico y de 15 y 10% mediante fluorocitometría. Los hallazgos inmunofenotípicos en los cuatro pacientes fueron variables (expresión constante de CD34, CD45, CD117 y CD7 y variable de CD13, CD33, CD64, CD11b, CD5, CD56 y HLA-DR). La expresión de marcadores de diferenciación megacariocítica (CD61, CD41, CD42) resultó también variable (tabla 1, fig. 2). El examen citogenético identificó la trisomía 21 en los cuatro casos (tabla 1) mostrando un paciente una doble población (mosaico). Una paciente desarrolló como anomalía adicional una trisomía 8 ausente en un control previo (tabla 1).
El análisis mutacional de GATA1 mostró en los 4 casos la presencia de una clona mutante en el exón 2 de dicho gen (tabla 1). La secuenciación genómica demostró una sustitución puntual en 3 pacientes en las posiciones 147, 266 y 161 (fig. 3A y B). En un caso el estudio de secuenciación resultó normal mientras que el perfil de la cromatografía mostró cambios sutiles sugestivos de la presencia de una pequeña clona GATA1 mutante (fig. 3C), lo que se atribuyó a la escasa proporción de células blásticas en el momento del diagnóstico1,21.
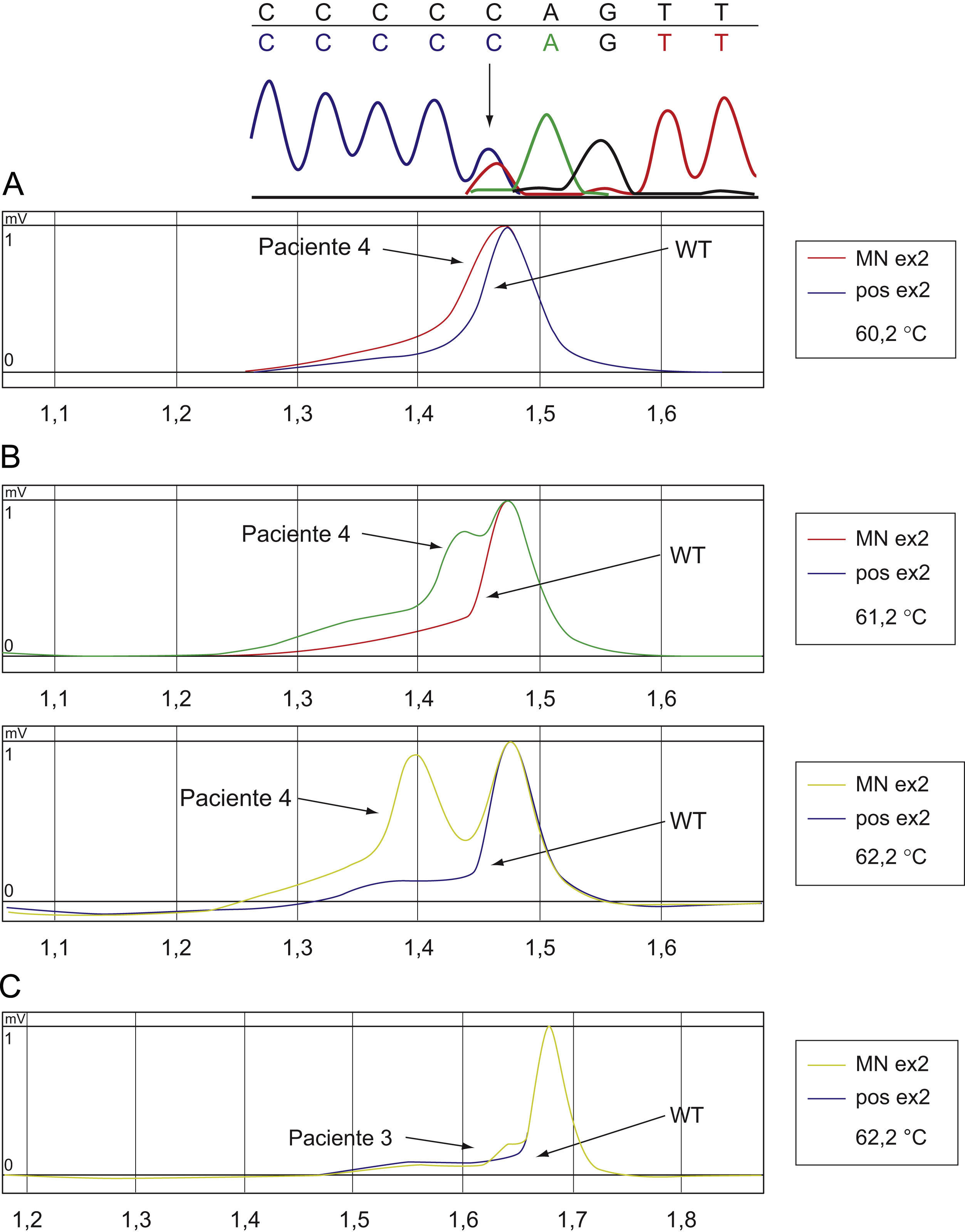
La secuenciación del gen GATA1 del paciente 4 (A) muestra una sustitución C>T (flecha) en la posición 161 dando lugar a un codón de parada. La cromatografía (B) muestra un patrón alternativo a GATA1 salvaje (WT: wild type). La cromatografía en el paciente 3 (C) muestra cambios sutiles sugestivos de la presencia de una pequeña clona GATA1 mutante.
Los dos casos de EMTN recibieron tratamiento de soporte mostrando normalización hematológica espontánea tras 60 y 100 días de seguimiento. En uno de ellos se comprobó la normalización espontánea en el análisis mutacional de GATA1 en un aspirado medular practicado a los 4 meses del diagnóstico. Los dos casos de LAMK se trataron según el protocolo «AML-BFM 98» en su rama adaptada para niños con SD11. La primera paciente se encuentra fuera de tratamiento y en remisión completa tras un seguimiento de 36 meses, habiéndose comprobado la remisión molecular en el análisis mutacional de GATA1 realizado a los 3 y 12 meses de finalizar su tratamiento. La segunda paciente falleció a consecuencia de una neumonía intersticial tras la terapia de inducción (tabla 1).
DiscusiónLa LAMK del SD constituye por varios motivos una entidad única3,11. Es el subtipo de leucemia aguda mieloblástica predominante en niños con SD: más de 90% de los casos frente a un 6% en el resto de niños. La edad de presentación es inferior a la edad en que se suele diagnosticar la leucemia aguda mieloblástica en el resto de niños: 2 años frente a 8 años5,6,13. La LAMK de los niños con SD suele venir precedida por un SMD hasta en un 62% de casos, hecho que sólo se documenta en 8% de pacientes sin SD con leucemia aguda mieloblástica5,17,22. Pero la peculiaridad más llamativa de la LAMK del SD es su asociación a la EMTN, un trastorno hematológico característico de recién nacidos con SD. Se estima que entre 4 y 10% de neonatos con SD tienen una proliferación clonal de megacarioblastos circulantes que resultan indistinguibles de los blastos de la LAMK1–4,16,17,23.
El proceso que da lugar a la EMTN comienza en la vida intrauterina y en la mayoría de casos no produce ningún problema al recién nacido aunque en algunos casos, como consecuencia de la afectación visceral, este trastorno puede tener consecuencias potencialmente fatales1–4,17. Esta población de células atípicas desaparece espontáneamente durante los 3primeros meses de vida sin intervención terapéutica alguna, pero un 20–25% de casos desarrollarán un SMD y una LAMK durante los primeros cuatro años1,2,4,7,16. Diferentes hipótesis intentan explicar el curso autolimitado de la EMTN interpretándose como un trastorno propio de la hematopoyesis hepática fetal2,3. La identificación de mutaciones adquiridas específicas de GATA1 en la EMTN y la LAMK demuestra que la población leucémica de la LAMK procede de una clona de la EMTN1,6.
El gen GATA1 está ubicado en el cromosoma X y codifica un factor de transcripción hematopoyético cuya función resulta esencial para la maduración de los precursores eroitroides y megacariocíticos1,10. Las mutaciones de GATA1 descritas en los blastos leucémicos del SD se agrupan en el exón 2 y dan lugar a la expresión exclusiva de una isoforma acortada de GATA1 de 40-kD que carece del dominio N-terminal, conocida como GATA-1s (GATA-1 short)1–3,6,8,16,18,21,24,25. En ausencia de la trisomía 21, estas mutaciones en GATA1 no desencadenan leucemia aunque sí son capaces de alterar la hematopoyesis normal3,26. Tales mutaciones no son detectables cuando la leucemia o la EMTN alcanzan la remisión y, exceptuando un caso27, no se han encontrado en los blastos leucémicos de casos de LAMK no relacionados con el SD1,3,18,24,25,28.
Por tanto, la EMTN y la LAMK se interpretan como diferentes manifestaciones de un trastorno común. En la fase inicial de la EMTN, uno o varios clones de blastos megacariocíticos se acumularían en la sangre, hígado y médula ósea. Mecanismos de muerte celular programada, senescencia o diferenciación celular permitirían la resolución espontánea de la EMTN con la consiguiente extinción de las clonas leucémicas. La desaparición espontánea de estas células tras el nacimiento sugiere que existe un factor ambiental que resulta esencial para la proliferación de las mismas y que vendría determinado por el ambiente hematopoyético hepático fetal1,18,25. En un grupo de pacientes, sin embargo, una subclona latente daría lugar al trastorno clonal conocido como SMD, que evolucionaría invariablemente a LAMK. Las mutaciones de GATA1 actuarían como evento iniciador de esta secuencia patogénica. Se desconocen los factores genéticos o epigenéticos adicionales responsables de la transformación en SMD y LAMK1–3,17–19,25. Se ha especulado sobre la posibilidad de tratar a los pacientes con EMTN, incluyendo aquellos con enfermedad silente detectada en el análisis mutacional de GATA1, con dosis bajas de arabinósido de citosina para prevenir la evolución a SMD y LAMK y estas iniciativas constituirán objetivos de investigación a corto plazo3,16.
El diagnóstico de LAMK en estos pacientes suele establecerse con un porcentaje de blastos en médula ósea inferior al habitual en un debut de leucosis aguda8,14. En nuestros dos pacientes de LAMK la estimación del porcentaje de blastos mediante citología y citometría osciló entre 10 y 48%. Tanto en la EMTN como en la LAMK los blastos leucémicos muestran rasgos variables de diferenciación megacariocítica (citológicos, fenotípicos y ultraestructurales)4,15, pero también se ha reconocido en ellos la expresión de características propias de otras líneas4–7,15,18. La expresión aberrante del marcador linfoide T CD7 (constante en nuestra serie) y de otros marcadores como CD56 en los blastos de la EMTN y la LAMK asociada al SD ya ha sido descrita previamente4,8,15,29 La escasa infiltración medular al debut y la variabilidad fenotípica de los blastos leucémicos podrían contribuir a dificultar y retrasar el diagnóstico de LAMK y en aquellos casos en los que el examen convencional de la médula ósea resulta equívoco, el examen molecular de GATA1 puede ser concluyente20.
En nuestra experiencia, el análisis mutacional de GATA1 ha resultado de gran utilidad. En primer lugar, ha permitido establecer con fidelidad el diagnóstico de trastorno mieloproliferativo asociado a SD, facilitando la orientación terapéutica: actitud expectante en los dos casos de MAT y tratamiento «adaptado» en los dos casos de LAMK. En segundo lugar, ha permitido «adaptar» la intensidad de tratamiento a una paciente de LAMK fenotípicamente normal portadora de un mosaicismo para la trisomía 21, reduciendo por tanto el riesgo de toxicidad sin comprometer la efectividad del tratamiento. En nuestra opinión, la recomendación de manejar la EMTN asociada a mutaciones en GATA1 en individuos portadores de mosaicismo21,30 de igual modo que la EMTN del SD debería extenderse también para el tratamiento de los pacientes con LAMK con mutaciones de GATA1 y mosaicismo 21, reduciendo por tanto su intensidad de tratamiento1.
Por último, el examen de GATA1 ha permitido establecer la remisión molecular en dos pacientes supervivientes en los que el análisis de enfermedad residual se veía comprometido por los fenotipos inespecíficos de las poblaciones blásticas3,20.
La LAMK de los niños con SD menores de 5 años es una entidad biológica única de pronóstico favorable cuyo tratamiento debe individualizarse del resto de casos de leucemia aguda mieloblástica. La identificación de las mutaciones específicas de GATA1 debe contribuir a identificar estos casos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
