



El síndrome de Apert (OMIM #101200), consistente en una anomalía craneofacial denominada acrocefalosindactilia tipo i, produce malformaciones en cráneo, cara, manos y pies, además de diversas alteraciones funcionales, como aumento de la presión intracraneal, problemas cardiorrespiratorios, deficiencia mental, ceguera, pérdida de la audición y otitis, entre otras1,2. Baumgartner, en 1842, y Wheaton, en 1894, hacen las primeras menciones sobre este síndrome. Sin embargo, fue el médico francés Eugene Apert quien lo describe y publica en detalle en 1906. Representa el 4,5% de todos los casos de craneosinostosis, con una distribución por sexo de 1:1 y una prevalencia en nuestro ámbito de 0,004/10.000 nacimientos3. Se hereda de forma autosómica dominante, aunque la mayoría de los casos se deben a mutaciones de novo en parejas no afectadas, incrementándose el riesgo con la edad paterna4. En esta afección, durante el desarrollo embrionario, el cráneo se fusiona prematuramente impidiendo su crecimiento normal, el tercio medio de la cara está hipoplásico, dando un aspecto de hundimiento del mismo, y las manos y los pies presentan una fusión entre los dedos, la cual puede variar en cuanto a involucrar solamente los tejidos blandos o extenderse también a los huesos.
Mutaciones en el gen FGPFR2 producen un incremento en el número de células precursoras que entran en la ruta osteogénica, lo que origina un aumento en la formación de matriz ósea subperióstica y una osificación prematura de la calota durante el desarrollo fetal5. Miraoui et al.6, mediante análisis de microarray para investigar las rutas de señalización que se activan por mutaciones en el gen FGFR2, descubrieron que se producía un aumento en la expresión de los genes EGFR y PDGRF-alfa.
Describimos el caso esporádico, sin antecedentes familiares, de un feto femenino en la semana 18, con anomalías craneales y sindactilia en manos y pies, detectadas mediante ecografía. En el seguimiento ecográfico en la semana 20 se objetiva hidronefrosis leve bilateral y falta de separación de los dedos. Cráneo en fresa, con estructuras intracraneales aparentemente normales. En la semana 21 se visualizan falanges, sin apertura de los dedos. En el corte coronal se visualiza imagen compatible con craneosinostosis. En la semana 22+4: craneosinostosis, abombamiento frontal, hipertelorismo y orejas de implantación baja. Sindactilia de los dedos de ambas manos. Las malformaciones indican un síndrome de Apert y los padres deciden una interrupción voluntaria del embarazo.
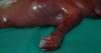
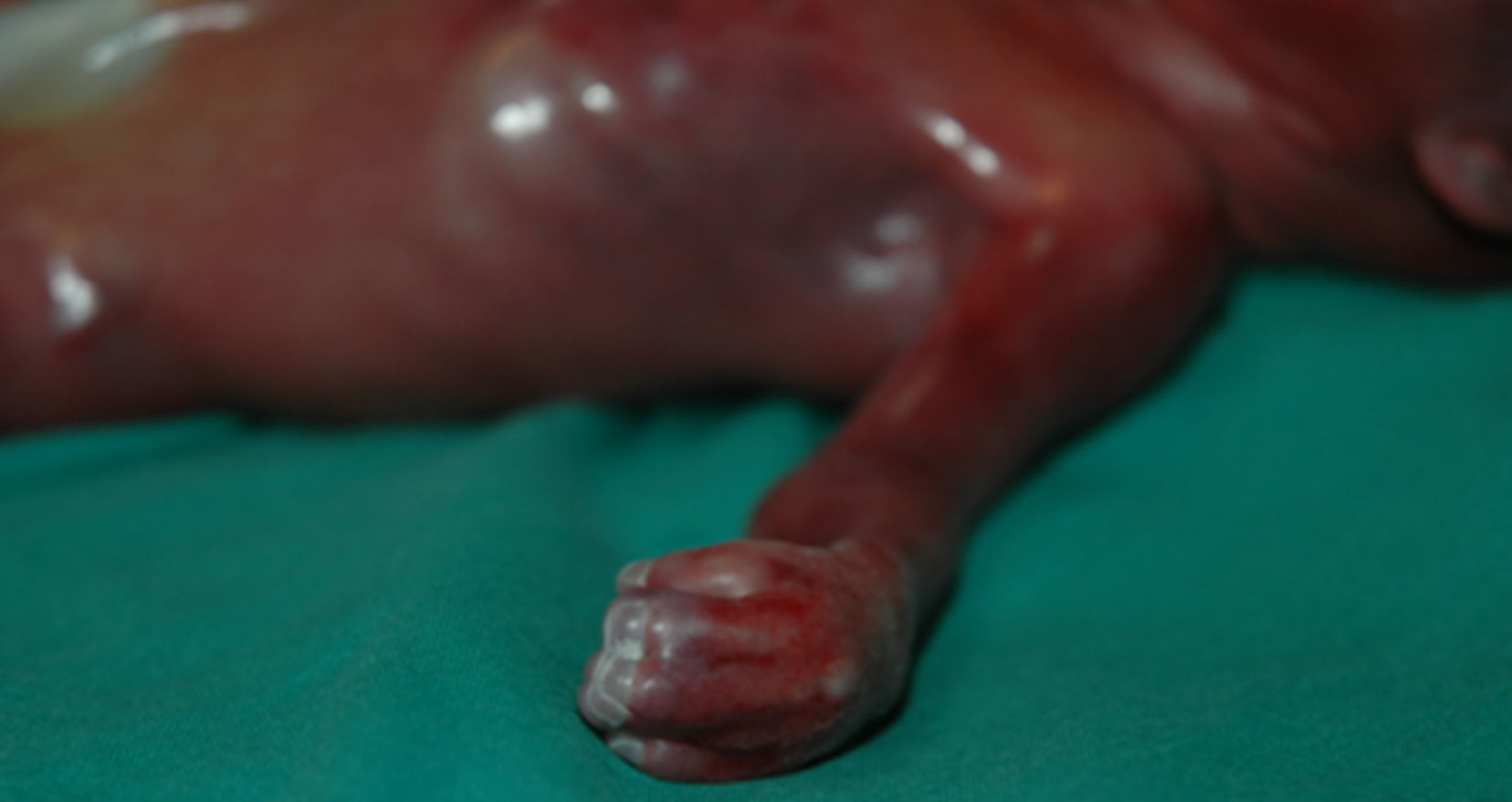
El estudio anatomopatológico describe a un feto de sexo femenino de 22 semanas con síndrome polimalformativo, peso 500 g, 27,5cm de longitud, que presenta unas proporciones corporales conservadas. Externamente se observa craneosinostosis de la sutura coronal bilateral, con fontanela anterior amplia, dehiscencia de la sutura metópica, sinostosis de la sutura lambdoidea y fontanela posterior permeable. En la cara se observa proptosis ocular, leve hipertelorismo e implantación baja de orejas. En la cavidad oral existe una fisura palatina medial, en forma de u invertida que comprende paladar blando y el tercio posterior del paladar duro, sin afectación de labios ni de encías. Los dedos de las manos y los pies presentan braquidactilia, pulgares y primeros dedos de los pies gruesos. Pulgares con desviación radial. Sindactilia cutánea 2-3-4-5 dedos de las manos y los pies. Palmas cóncavas. Espalda, tórax y abdomen, normales. Cordón trivascular. Lecho ungueal continuo en 2-3-4-5 de las manos. Ano en posición normal. Genitales normoconformados de sexo femenino (fig. 1).
Con la sospecha clínica de síndrome de Apert se realiza un estudio genético a partir de ADN de tejido fetal del exón 7 del gen FGFR2, donde se localizan los codones 252 y 253, descritos como causantes de esta enfermedad mediante PCR y posterior secuenciación7. El feto era portador en heterocigosis de la mutación: c.755C>G, p.Ser252Trp (NM_022970) (fig. 2). Las mutaciones en los codones 252 y 253 parecen ser de origen paterno, asociadas a edad mayor de 35 años8. En el caso que nos ocupa, el padre tenía 41 años. Este resultado confirma el diagnóstico clínico de síndrome de Apert.
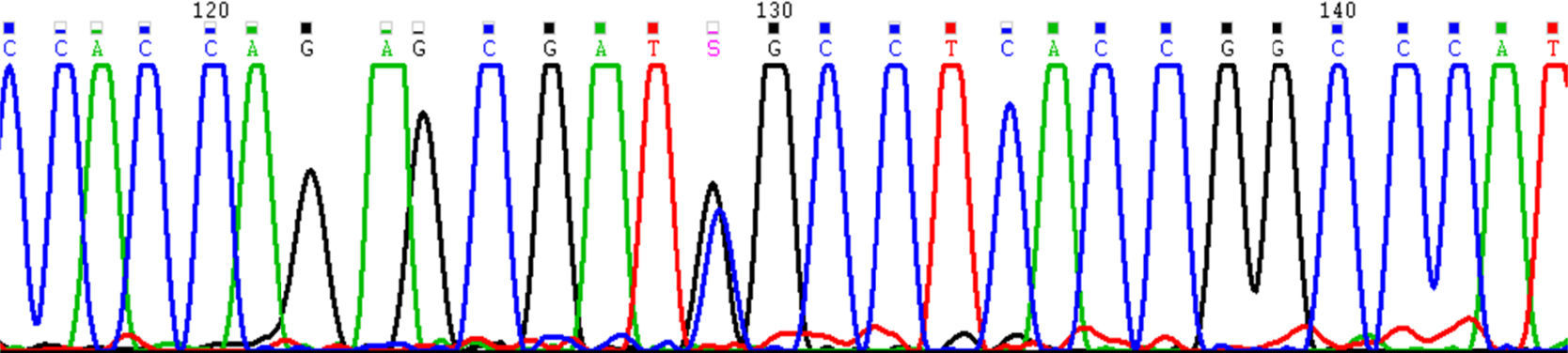
La amplificación y posterior secuenciación del exón 7 del gen FGFR2 detectó la presencia en heterocigosis de la mutación S252W (c.755C>G). Esta mutación, junto con la del codón adyacente P253R (c.758C>G), son las responsables de la aparición del síndrome de Apert. Mutaciones germinales en el gen FGFR2 producen alteraciones esqueléticas congénitas debido a una activación anómala de las rutas de transducción de señales en las que participa.
Son muy escasos los casos diagnosticados de síndrome de Apert en el segundo trimestre del embarazo, excluyendo aquellos publicados de madres afectadas o en el contexto de recurrencia9,10. El diagnóstico prenatal ecográfico se puede confirmar mediante la realización de una prueba genética que demuestre la mutación en el gen FGFR2 en el líquido amniótico o la vellosidad corial. Este tipo de síndrome exhibe cariotipos normales y los niveles de alfafetoproteína también se encuentran dentro de los límites normales, por lo que no resultan útiles los sistemas de cribado prenatal de cromosomopatías.
