

Las cardiopatías congénitas (CC) son los defectos congénitos (DC) más comunes.
ObjetivoConocer la prevalencia total de las CC en Asturias y su tendencia y realizar una descripción de las anomalías asociadas y los síndromes o las secuencias.
Material y métodosAnálisis de los datos del Registro de DC de Asturias de los años 1990–2004. La población estudio fueron los 103.452 nacidos de madres residentes en Asturias en ese período. Se calcularon las tasas de prevalencia total y al nacimiento.
ResultadosDe los 3.035 casos con DC registrados durante los 15 años estudiados, 778 tenían una CC. La prevalencia total media fue de 75,2 por 10.000 nacidos, con una tendencia ascendente. Las más frecuentes fueron la comunicación interventricular (28,8 por cada 10.000 nacidos vivos), los defectos del septo auricular (10,3 por cada 10.000 nacidos vivos) y la persistencia del ductus arterioso (6,0 por cada 10.000 nacidos vivos). El 73,6% de las CC se presentó de forma aislada, el 12,5% asociadas a otras anomalías congénitas y el 14% pertenecía a un síndrome o a una secuencia. El diagnóstico prenatal fue del 7,3% (del 3,8% en los casos aislados).
ConclusionesLa prevalencia total de las CC en Asturias durante este período fue similar a la de otros registros europeos. El aparente incremento de la prevalencia se debió a un mayor diagnóstico de los defectos menores, mientras que las CC más graves mantuvieron una frecuencia estable. El diagnóstico prenatal de las CC en Asturias fue inferior al de otros registros europeos.
Congenital heart diseases (CHDs) are the most common type of birth defect.
ObjectiveThe purpose of this investigation was to assess the prevalence and trends of CHDs, and to describe the associated malformations and syndromes or sequences in a geographically defined population.
Material and methodsData wers collected from the Asturias Registry of Congenital Defects. The period studied was from 1990 to 2004, and the study population was the 103,452 births of mothers living in the region. Total prevalence and birth prevalence were calculated.
ResultsA total of 3035 cases with congenital defects were recorded, of these 778 had CHDs. The total prevalence was 75.2 per 10000 births, with an upward trend during this period. The most common CHDs were: ventricular septal defects (28.8 per 10000 births), atrial septal defects (10.3 per 10000 births) and patent ductus arteriosus (6.0 per 10000 births). A total of 73.6% of CHDs occurred as isolated defects, 12.5% with other congenital defects and 14% were syndromes or sequences. Prenatal diagnosis was effective in only 7.3% (3.8% in isolated cases).
ConclusionsThe prevalence of CHDs in Asturias over this period falls within the range reported for other European registries. The apparent increase in prevalence of CHD results mainly from improved diagnosis of minor defects, but there has been no change over time in birth prevalence of more serious defects.
Las cardiopatías congénitas (CC) son los defectos congénitos (DC) más comunes, con una frecuencia estimada al nacimiento de entre 5,2–12,5 por cada 1.000 nacidos1–4. Esta elevada variabilidad se debe a múltiples factores, entre los que destacan los criterios de inclusión, la capacidad diagnóstica, el límite de edad para el momento del diagnóstico, etc. Las CC constituyen un grupo de DC muy heterogéneo, y se presentan en numerosas ocasiones más de un tipo de defecto cardíaco en un mismo paciente. Las CC, tanto aisladas como asociadas a otros DC o síndromes, son una causa importante de morbimortalidad fundamentalmente en la etapa perinatal.
El objetivo de este estudio fue conocer la frecuencia de las CC en Asturias, así como su evolución, durante los años 1990–2004 y realizar una descripción clinicoepidemiológica de éstas.
Material y métodosEl Registro de DC de Asturias (RDCA) es un registro de base poblacional, es decir, pretende la captación de todos los casos de DC (nacidos vivos, mortinatos y abortos inducidos [AI]) de madres residentes en Asturias. Las características del RDCA5 están ampliamente descritas y recogidas en la página web del EUROCAT (European Concerted Action on Congenital Anomalies and Twins)6, con el que comparte, en lo fundamental, una metodología común.
Los 7 hospitales públicos de Asturias (Jarrio, Carmen y Severo Ochoa, San Agustín, Universitario Central de Asturias, Cabueñes, Álvarez-Buylla y Valle del Nalón) forman parte del RDCA desde su puesta en marcha en el año 1990, y posteriormente se incorporó el Hospital de Oriente tras su apertura en 1996.
Las fuentes de información del RDCA son los servicios de Pediatría/Neonatología y Obstetricia, de Genética Clínica y la Unidad de Diagnóstico Prenatal, los servicios de Anatomía Patológica, los de Archivos e Historias Clínicas y los de Atención al Paciente; el Registro del Programa de Detección Neonatal de Hipotiroidismo Congénito y Fenilcetonuria, el Registro del Programa de Detección Prenatal de los Defectos del Tubo Neural, el Registro de Interrupciones Voluntarias del Embarazo y el Registro de Mortalidad de la Consejería de Salud y Servicios Sanitarios.
Los pediatras recogen de forma sistemática los casos de DC y los envían mensualmente al registro, y se incluyen en la base de datos una vez completados (si es preciso, se requiere información adicional). Anualmente, con el fin de completar el proceso de captación, se realiza una búsqueda activa de casos al cruzar la base de datos de los casos ya captados con el resto de fuentes de información ya mencionadas. El mayor número de casos nuevos, en general, y específicamente en el caso de las CC que se diagnostican más allá del período neonatal, lo aporta el Servicio de Archivos e Historias Clínicas, a través del sistema del conjunto mínimo básico de datos, común a todos los hospitales y que se refiere a los diagnósticos en el momento del alta hospitalaria del caso.
El RDCA realiza un sistema de vigilancia y captación de casos diagnosticados hasta los 5 años de vida. Los casos de DC registrables incluyen los definidos por el EUROCAT y excluye las anomalías menores y aisladas también recogidas en su manual operacional. El EUROCAT es un grupo de trabajo constituido por registros europeos de DC de base poblacional de 20 países, incluido el RDCA.
El grupo de trabajo del RDCA está constituido por pediatras, genetistas y una epidemióloga de la Consejería de Salud y Servicios Sanitarios. La Consejería de Salud y Servicios Sanitarios y el Servicio de Salud del Principado de Asturias financian el RDCA.
El período de estudio fue del 1 de enero de 1990 al 31 de diciembre de 2004. Los casos de CC se extrajeron de la base de datos del RDCA mediante los códigos de la Clasificación Internacional de Enfermedades (CIE-9) (hasta el año 2000) y de la CIE-10 (desde el año 2001), tanto de nacidos vivos o muertos como de AI por DC. De ellos, se excluyeron las malposiciones cardíacas sin anomalías estructurales y las arritmias. Los casos con más de una CC se clasificaron por la más grave.
Para el análisis de la frecuencia se utilizó como indicador principal la tasa de prevalencia total: total de casos (vivos, muertos y AI) registrados por cada 10.000 nacidos (vivos o muertos). En los defectos susceptibles de diagnóstico prenatal y posterior interrupción del embarazo, se analizó además la tasa de prevalencia al nacimiento, que no incluye en el numerador a los AI. El cálculo de los intervalos de confianza de las tasas de prevalencia se realizó mediante la fórmula reducida del error estándar: 1,96*raíz (p*q/N) (N es el número de nacidos en Asturias en el período estudiado). El porcentaje de diagnóstico prenatal se calculó referente a los casos aislados, ya que parte del global corresponde con mucha probabilidad al diagnóstico prenatal de otros defectos asociados a las cardiopatías.
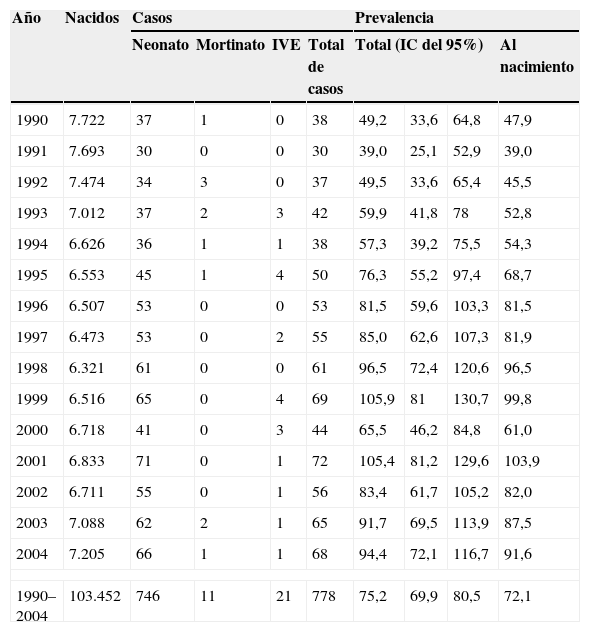
ResultadosDe los 3.035 casos con DC registrados por el RDCA durante los 15 años estudiados, 778 tenían una CC; el corazón constituyó el órgano más afectado por DC y presentó una prevalencia total media de 75,2 por cada 10.000 nacidos. Pero esta tasa no fue estable, sino con una tendencia ascendente. La tabla 1 presenta la prevalencia total y al nacimiento, y el impacto del diagnóstico prenatal resulta la diferencia entre ellas.
Frecuencia y evolución de las cardiopatías congénitas. Asturias, 1990–2004
| Año | Nacidos | Casos | Prevalencia | ||||||
| Neonato | Mortinato | IVE | Total de casos | Total (IC del 95%) | Al nacimiento | ||||
| 1990 | 7.722 | 37 | 1 | 0 | 38 | 49,2 | 33,6 | 64,8 | 47,9 |
| 1991 | 7.693 | 30 | 0 | 0 | 30 | 39,0 | 25,1 | 52,9 | 39,0 |
| 1992 | 7.474 | 34 | 3 | 0 | 37 | 49,5 | 33,6 | 65,4 | 45,5 |
| 1993 | 7.012 | 37 | 2 | 3 | 42 | 59,9 | 41,8 | 78 | 52,8 |
| 1994 | 6.626 | 36 | 1 | 1 | 38 | 57,3 | 39,2 | 75,5 | 54,3 |
| 1995 | 6.553 | 45 | 1 | 4 | 50 | 76,3 | 55,2 | 97,4 | 68,7 |
| 1996 | 6.507 | 53 | 0 | 0 | 53 | 81,5 | 59,6 | 103,3 | 81,5 |
| 1997 | 6.473 | 53 | 0 | 2 | 55 | 85,0 | 62,6 | 107,3 | 81,9 |
| 1998 | 6.321 | 61 | 0 | 0 | 61 | 96,5 | 72,4 | 120,6 | 96,5 |
| 1999 | 6.516 | 65 | 0 | 4 | 69 | 105,9 | 81 | 130,7 | 99,8 |
| 2000 | 6.718 | 41 | 0 | 3 | 44 | 65,5 | 46,2 | 84,8 | 61,0 |
| 2001 | 6.833 | 71 | 0 | 1 | 72 | 105,4 | 81,2 | 129,6 | 103,9 |
| 2002 | 6.711 | 55 | 0 | 1 | 56 | 83,4 | 61,7 | 105,2 | 82,0 |
| 2003 | 7.088 | 62 | 2 | 1 | 65 | 91,7 | 69,5 | 113,9 | 87,5 |
| 2004 | 7.205 | 66 | 1 | 1 | 68 | 94,4 | 72,1 | 116,7 | 91,6 |
| 1990–2004 | 103.452 | 746 | 11 | 21 | 778 | 75,2 | 69,9 | 80,5 | 72,1 |
IC: intervalo de confianza; IVE: interrupción voluntaria del embarazo.
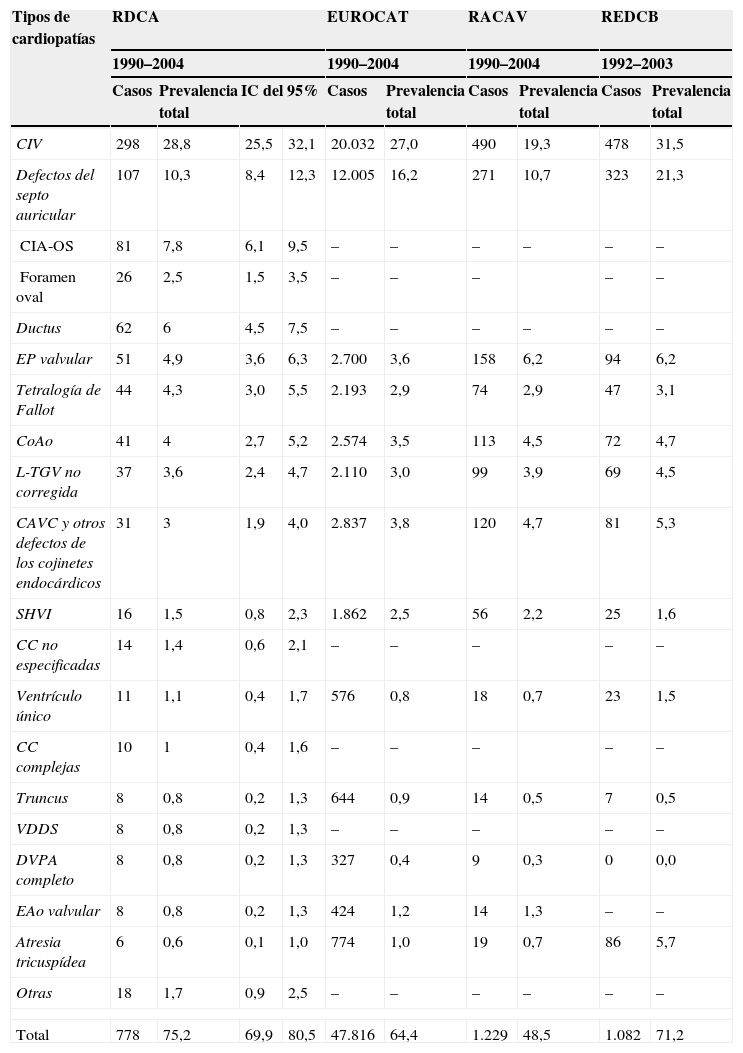
En la tabla 2 se muestra la frecuencia de los distintos tipos de CC en Asturias comparada con otros registros españoles de base poblacional y con el EUROCAT. La comunicación interventricular (CIV) fue la más frecuente, seguida de los defectos del septo auricular (Def.SA), fundamentalmente la comunicación interauricular óstium secúndum (CIA-OS) y la persistencia del ductus arterioso. La suma de estos 467 casos supone más del 60% de todos los casos. La evolución de la frecuencia del conjunto de estos 3 defectos mostró una tendencia a aumentar mayor que el total de las CC, mientras el grupo del resto de las CC tuvo una frecuencia estable durante el período estudiado (fig. 1).
Frecuencia de los distintos tipos de cardiopatías congénitas en Asturias y comparación con otros registros españoles de base poblacional y con el European Concerted Action on Congenital Anomalies and Twins
| Tipos de cardiopatías | RDCA | EUROCAT | RACAV | REDCB | ||||||
| 1990–2004 | 1990–2004 | 1990–2004 | 1992–2003 | |||||||
| Casos | Prevalencia total | IC del 95% | Casos | Prevalencia total | Casos | Prevalencia total | Casos | Prevalencia total | ||
| CIV | 298 | 28,8 | 25,5 | 32,1 | 20.032 | 27,0 | 490 | 19,3 | 478 | 31,5 |
| Defectos del septo auricular | 107 | 10,3 | 8,4 | 12,3 | 12.005 | 16,2 | 271 | 10,7 | 323 | 21,3 |
| CIA-OS | 81 | 7,8 | 6,1 | 9,5 | – | – | – | – | – | – |
| Foramen oval | 26 | 2,5 | 1,5 | 3,5 | – | – | – | – | – | |
| Ductus | 62 | 6 | 4,5 | 7,5 | – | – | – | – | – | – |
| EP valvular | 51 | 4,9 | 3,6 | 6,3 | 2.700 | 3,6 | 158 | 6,2 | 94 | 6,2 |
| Tetralogía de Fallot | 44 | 4,3 | 3,0 | 5,5 | 2.193 | 2,9 | 74 | 2,9 | 47 | 3,1 |
| CoAo | 41 | 4 | 2,7 | 5,2 | 2.574 | 3,5 | 113 | 4,5 | 72 | 4,7 |
| L-TGV no corregida | 37 | 3,6 | 2,4 | 4,7 | 2.110 | 3,0 | 99 | 3,9 | 69 | 4,5 |
| CAVC y otros defectos de los cojinetes endocárdicos | 31 | 3 | 1,9 | 4,0 | 2.837 | 3,8 | 120 | 4,7 | 81 | 5,3 |
| SHVI | 16 | 1,5 | 0,8 | 2,3 | 1.862 | 2,5 | 56 | 2,2 | 25 | 1,6 |
| CC no especificadas | 14 | 1,4 | 0,6 | 2,1 | – | – | – | – | – | |
| Ventrículo único | 11 | 1,1 | 0,4 | 1,7 | 576 | 0,8 | 18 | 0,7 | 23 | 1,5 |
| CC complejas | 10 | 1 | 0,4 | 1,6 | – | – | – | – | – | |
| Truncus | 8 | 0,8 | 0,2 | 1,3 | 644 | 0,9 | 14 | 0,5 | 7 | 0,5 |
| VDDS | 8 | 0,8 | 0,2 | 1,3 | – | – | – | – | – | |
| DVPA completo | 8 | 0,8 | 0,2 | 1,3 | 327 | 0,4 | 9 | 0,3 | 0 | 0,0 |
| EAo valvular | 8 | 0,8 | 0,2 | 1,3 | 424 | 1,2 | 14 | 1,3 | – | – |
| Atresia tricuspídea | 6 | 0,6 | 0,1 | 1,0 | 774 | 1,0 | 19 | 0,7 | 86 | 5,7 |
| Otras | 18 | 1,7 | 0,9 | 2,5 | – | – | – | – | – | – |
| Total | 778 | 75,2 | 69,9 | 80,5 | 47.816 | 64,4 | 1.229 | 48,5 | 1.082 | 71,2 |
CAVC: canal auriculoventricular común; CC: cardiopatías congénitas; CIA-OS: comunicación interauricular óstium secúndum; CIV: comunicación interventricular; CoAo: coartación de la aorta; DVPA: drenaje venoso pulmonar anómalo; EAo: estenosis aórtica; EP: estenosis pulmonar; EUROCAT: European Concerted Action on Congenital Anomalies and Twins; IC: intervalo de confianza; L-TGV: L-transposición de los grandes vasos; RACAV: Registro de Anomalías Congénitas de la Comunidad Autónoma del País Vasco; RDCA: Registro de Defectos Congénitos de Asturias; REDCB: Registro de Defectos Congénitos de Barcelona; SHVI: síndrome de hipoplasia del ventrículo izquierdo; VDDS: ventrículo derecho de doble salida.

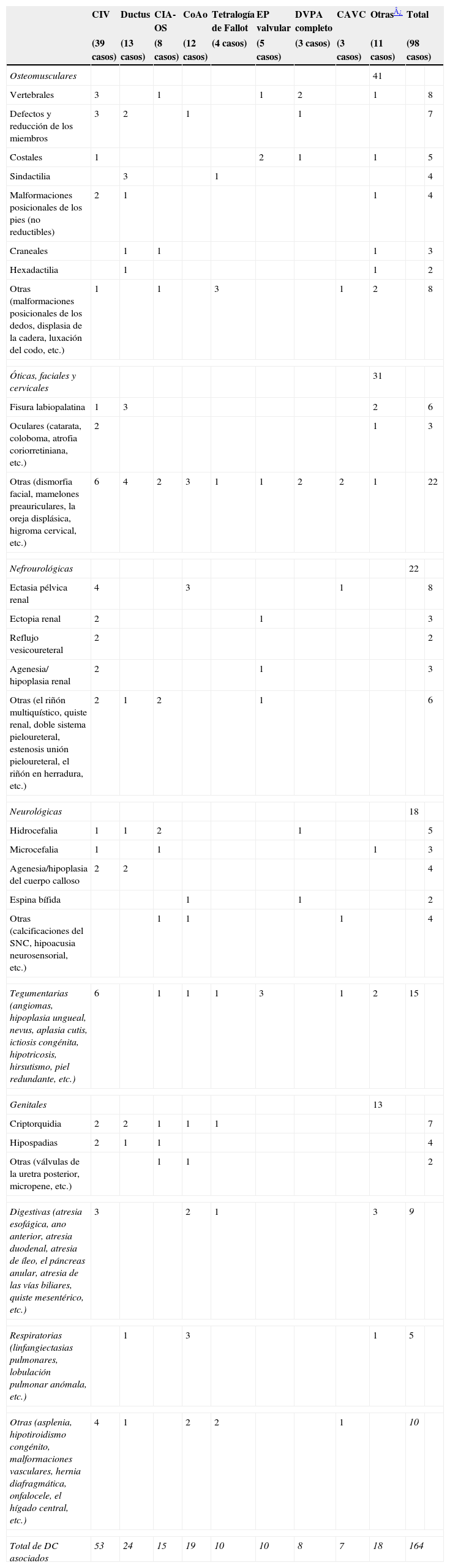
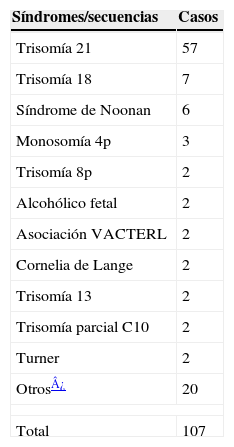
El 73,6% de las CC se presentó de forma aislada. La tabla 4 refleja los casos pertenecientes a síndromes y a asociaciones de alta frecuencia (13,7%). Entre ellos, el predominante fue la trisomía 21, que además fue la única entidad asociada a los defectos de los cojinetes endocárdicos (completos o incompletos). El 12,6% restante de CC, sin formar parte de un síndrome, tenía otra/s anomalía/s asociada/s, muy heterogéneas, que se detallan en la tabla 3. Del total de CIV, Def.SA y ductus, el 7,5% estaba asociado a otro DC (62 casos), mientras que el 11,2% (35 casos) lo estaba del resto de las CC. La única secuencia registrada fue un caso de Potter, que se incluyó en este grupo.
Distribución de las anomalías asociadas según el tipo de cardiopatía congénita (se excluyen los síndromes y las asociaciones de alta frecuencia). Asturias, 1990–2004
| CIV | Ductus | CIA-OS | CoAo | Tetralogía de Fallot | EP valvular | DVPA completo | CAVC | Otras¿ | Total | ||
| (39 casos) | (13 casos) | (8 casos) | (12 casos) | (4 casos) | (5 casos) | (3 casos) | (3 casos) | (11 casos) | (98 casos) | ||
| Osteomusculares | 41 | ||||||||||
| Vertebrales | 3 | 1 | 1 | 2 | 1 | 8 | |||||
| Defectos y reducción de los miembros | 3 | 2 | 1 | 1 | 7 | ||||||
| Costales | 1 | 2 | 1 | 1 | 5 | ||||||
| Sindactilia | 3 | 1 | 4 | ||||||||
| Malformaciones posicionales de los pies (no reductibles) | 2 | 1 | 1 | 4 | |||||||
| Craneales | 1 | 1 | 1 | 3 | |||||||
| Hexadactilia | 1 | 1 | 2 | ||||||||
| Otras (malformaciones posicionales de los dedos, displasia de la cadera, luxación del codo, etc.) | 1 | 1 | 3 | 1 | 2 | 8 | |||||
| Óticas, faciales y cervicales | 31 | ||||||||||
| Fisura labiopalatina | 1 | 3 | 2 | 6 | |||||||
| Oculares (catarata, coloboma, atrofia coriorretiniana, etc.) | 2 | 1 | 3 | ||||||||
| Otras (dismorfia facial, mamelones preauriculares, la oreja displásica, higroma cervical, etc.) | 6 | 4 | 2 | 3 | 1 | 1 | 2 | 2 | 1 | 22 | |
| Nefrourológicas | 22 | ||||||||||
| Ectasia pélvica renal | 4 | 3 | 1 | 8 | |||||||
| Ectopia renal | 2 | 1 | 3 | ||||||||
| Reflujo vesicoureteral | 2 | 2 | |||||||||
| Agenesia/ hipoplasia renal | 2 | 1 | 3 | ||||||||
| Otras (el riñón multiquístico, quiste renal, doble sistema pieloureteral, estenosis unión pieloureteral, el riñón en herradura, etc.) | 2 | 1 | 2 | 1 | 6 | ||||||
| Neurológicas | 18 | ||||||||||
| Hidrocefalia | 1 | 1 | 2 | 1 | 5 | ||||||
| Microcefalia | 1 | 1 | 1 | 3 | |||||||
| Agenesia/hipoplasia del cuerpo calloso | 2 | 2 | 4 | ||||||||
| Espina bífida | 1 | 1 | 2 | ||||||||
| Otras (calcificaciones del SNC, hipoacusia neurosensorial, etc.) | 1 | 1 | 1 | 4 | |||||||
| Tegumentarias (angiomas, hipoplasia ungueal, nevus, aplasia cutis, ictiosis congénita, hipotricosis, hirsutismo, piel redundante, etc.) | 6 | 1 | 1 | 1 | 3 | 1 | 2 | 15 | |||
| Genitales | 13 | ||||||||||
| Criptorquidia | 2 | 2 | 1 | 1 | 1 | 7 | |||||
| Hipospadias | 2 | 1 | 1 | 4 | |||||||
| Otras (válvulas de la uretra posterior, micropene, etc.) | 1 | 1 | 2 | ||||||||
| Digestivas (atresia esofágica, ano anterior, atresia duodenal, atresia de íleo, el páncreas anular, atresia de las vías biliares, quiste mesentérico, etc.) | 3 | 2 | 1 | 3 | 9 | ||||||
| Respiratorias (linfangiectasias pulmonares, lobulación pulmonar anómala, etc.) | 1 | 3 | 1 | 5 | |||||||
| Otras (asplenia, hipotiroidismo congénito, malformaciones vasculares, hernia diafragmática, onfalocele, el hígado central, etc.) | 4 | 1 | 2 | 2 | 1 | 10 | |||||
| Total de DC asociados | 53 | 24 | 15 | 19 | 10 | 10 | 8 | 7 | 18 | 164 | |
CAVC: canal auriculoventricular común; CC: cardiopatía congénita; CIA-OS: comunicación interauricular óstium secúndum; CIV: comunicación interventricular; CoAo: coartación de la aorta; DC: defecto congénito; DVPA: drenaje venoso pulmonar anómalo; EP: estenosis pulmonar; FO: foramen oval; HBV: hipoplasia biventricular; MCP: miocardiopatía; SHVI: síndrome de hipoplasia del ventrículo izquierdo; SNC: sistema nervioso central; VU: ventrículo único.
Síndromes y asociaciones de alta frecuencia registrados en los casos con cardiopatía congénita. Asturias, 1990–2004
| Síndromes/secuencias | Casos |
| Trisomía 21 | 57 |
| Trisomía 18 | 7 |
| Síndrome de Noonan | 6 |
| Monosomía 4p | 3 |
| Trisomía 8p | 2 |
| Alcohólico fetal | 2 |
| Asociación VACTERL | 2 |
| Cornelia de Lange | 2 |
| Trisomía 13 | 2 |
| Trisomía parcial C10 | 2 |
| Turner | 2 |
| Otros¿ | 20 |
| Total | 107 |
Otros: Adams-Oliver, Alagille, trisomía 11q, Apert, cardiodigital, cardiofaciocutáneo, Carpenter, CATCH 22: (cardiopatía, facies anómala, hipoplasia tímica, paladar hendido (cleft), hipocalcemia). Di George, Crouzon, Charge, Holt-Oram, Marfan, neurofibromatosis I, Opitz, Pierre Robin, rubeola congénita, sotos, translocación (5;6), y translocación (5;X): VACTERL: defectos vertebrales, atresia anorectal, anomalías cardíacas, fístula traqueoesofágica con atresia esofágica, anomalías renales y defectos en miembros superiores.
Respecto al sexo del nacido, se encontró un ligero predominio de varones (53,6%). Sin embargo, éste fue más marcado en el caso de las estenosis aórticas (el 62,5% de niños), mientras que el ductus y la CIA-OS fueron más frecuentes en las niñas (el 51,6 y el 54,1%, respectivamente).
Se registró un porcentaje de prematuridad (edad gestacional inferior a 37 semanas) del 14,3%.
El diagnóstico prenatal de las CC en los casos aislados fue del 3,8%.
DiscusiónLa prevalencia de las CC presenta una amplia variabilidad (de 52–125 por cada 10.000 nacidos)1–4. Esto está relacionado con diferentes factores7: los criterios de inclusión muy diferentes de las series publicadas (fundamentalmente en los referidos a los defectos cardíacos menores), los grandes avances tecnológicos no homogéneos durante este período, que permiten tanto el diagnóstico de defectos más leves (incluso indetectables de otra forma) como una mayor certeza diagnóstica, y por último, la distinta efectividad de la ecografía que depende de la capacidad y la experiencia de cada ecografista.
Teniendo todo esto en cuenta, la prevalencia total media de CC así como de los principales tipos de ellas en Asturias son similares a las de otros países de nuestro medio. Al igual que en ellos, la frecuencia registrada está aumentando de forma progresiva a expensas de los defectos cardíacos menores8 por una mayor sensibilidad y utilización de las pruebas diagnósticas, incluso en casos asintomáticos. Así, tras excluir los casos de CIV, los defectos del tabique interauricular y la persistencia del ductus arterioso, la prevalencia de CC se mostró estable durante estos 15 años en Asturias.
En relación con otros registros del EUROCAT, de similar metodología, nuestros resultados fueron similares a los del Registro de DC de Barcelona, sin diferencias relevantes respecto a la media de todos los registros del EUROCAT y con mayores frecuencias que las registradas por el Registro de Anomalías Congénitas de la Comunidad Autónoma del País Vasco, sobre todo debido a diferencias de registro en las CIV. Las diferencias al alza del RDCA también pueden explicarse por la inclusión de casos hasta los 5 años de vida, mientras que otros registros hacen seguimiento durante el primer año, y pueden incluir, por tanto, una mayor proporción de CC menores de detección más tardía. Nuestros datos no son comparables con los del Estudio Colaborativo Español de Malformaciones Congénitas (ECEMC) por su diferente metodología (recoge casos diagnosticados en los primeros 3 días de vida y no incluye los AI), ni tampoco, por el mismo motivo, con el estudio de Arias López (incluye sólo nacidos vivos y es retrospectivo). Al ser ambos de base hospitalaria, no incluyen hijos de madres residentes en el área de estudio, pero incluyen nacidos en otro lugar. Esto sería más improbable en el caso del ECEMC al estar incluidos hospitales de toda España, mientras que en el estudio de Badajoz se recaptarían aquellos que acudieran a consulta9.
Las CC mayores más frecuentes, la coartación de la aorta, el truncus y la L-transposición de los grandes vasos tuvieron durante este período en Asturias una prevalencia similar a la media del EUROCAT.
Respecto a las anomalías asociadas no cardiovasculares, aunque Pradat encontró algunas asociaciones específicas, como la transposición de grandes vasos y la atresia del esófago1, actualmente es un campo en estudio por la enorme variedad de tipos de CC, así como por su diferente etiología. En relación con los síndromes, está ampliamente descrita la asociación entre el síndrome de Down y los defectos de los cojinetes endocárdicos, también encontrada en este estudio, así como los defectos del tracto de salida y el CATCH (cardiopatía, facies anómala, hipoplasia tímica, paladar hendido [cleft], hipocalcemia) 22, la estenosis pulmonar y el síndrome de Noonan y LEOPARD (lunares o lentigines, anomalías de la conducción en electrocardiografía, hipertelorismo ocular/cardiomiopatía obstructiva, estenosis pulmonar, genitales anómalos, retraso en el crecimiento, sordera [deafness] sensoneural), la tetralogía de Fallot y el Alagille, etc10–13. Igual ocurre con la asociación de alta frecuencia VACTERL (defectos vertebrales, atresia anorectal, anomalías cardíacas, fístula traqueoesofágica con atresia esofágica, anomalías renales y defectos en miembros superiores), que incluye las CC dentro de su acrónimo14,15.
El diagnóstico prenatal de las CC aisladas en Asturias fue muy inferior al de otros registros del EUROCAT (alrededor del 10% en los casos aislados)16,17. Esto se puede deber, en parte, a un mayor rastreo de casos por el RDCA con la inclusión de CC más leves (por ejemplo, defectos del tabique sin limitación de tamaño ni gradiente). Nuestros pobres resultados también podrían estar influidos por la ausencia de un diagnóstico prenatal centralizado en las sospechas de CC con las características descritas por Allan: formación adecuada, mantenimiento continuo de capacidades con suficiente volumen de pacientes, incluyendo numerosos casos críticos, así como un continuo feedback18. Por otra parte, existe controversia sobre la conveniencia o no de efectuar ecocardiografía prenatal a todos los embarazos. Históricamente, sólo se realizaba en los casos con factores de riesgo18, pero ahora es habitual la evaluación de las 4 cámaras cardíacas al menos. Incluso Carvalho, en su estudio de 9.277 mujeres gestantes, defiende que el método más efectivo para la detección prenatal de CC es valorar el flujo de salida ventricular junto con la visualización de las 4 cámaras en la ecografía de rutina entre las 18 y las 23 semanas19.
Por otra parte, los grandes avances de la Cardiología Fetal probablemente faciliten mejoras en este campo. La ecografía tridimensional tiene una menor dependencia de la experiencia del ecografista, y permite mayor exactitud e incluso permite corregir algunos diagnósticos realizados por la ecografía bidimensional20,21.
La ausencia de un estudio necrópsico sistemático de los AI por DC en Asturias no permite una confirmación diagnóstica posterior así como una detección de otras anomalías asociadas (muy útil no sólo para la calidad del registro, sino también básico para el consejo genético sobre futuros embarazos de la madre). Tennstedt, en su análisis de necropsias, encontró que las CC estaban presentes en el 22% de los AI por detección prenatal de DC, el 9% de los abortos espontáneos y el 7% de los nacidos muertos22.
En conclusión, la prevalencia total media de las CC en Asturias fue de 75,2 casos por cada 10.000 nacidos, similar a la de otros países de nuestro medio. El aparente incremento de la prevalencia de las CC se debió a un mayor diagnóstico de los defectos menores, mientras que las CC más graves mantuvieron una frecuencia estable. El diagnóstico prenatal de las CC en Asturias fue inferior al de otros registros europeos.
La existencia de los registros de DC, en este caso el RDCA, es necesaria no sólo para la investigación epidemiológica, sino también para promover acciones formativas, planificar y evaluar políticas de diagnóstico prenatal y los recursos necesarios para favorecer con todo ello una mejor atención, ya desde antes de su nacimiento, a los niños con CC.
FinanciaciónConsejería de Salud y Servicios Sanitarios de Asturias.
Este trabajo ha sido posible gracias a las gerencias y a los trabajadores de los hospitales públicos del Principado de Asturias (Hospital de Jarrio, Hospital Carmen y Severo Ochoa, Hospital San Agustín, Hospital Universitario Central de Asturias, Hospital de Cabueñes, Hospital Grande Covián, Hospital Valle del Nalón, y Hospital Álvarez Buylla), que han colaborado en la recogida de datos y han facilitado en todo momento nuestra labor.
