




Conocer la prevalencia en España de los diferentes errores congénitos del metabolismo que presentan homocistinuria y establecer las medidas oportunas para garantizar su prevención, diagnóstico y tratamiento, en aquellos casos posibles.
Material y métodosEn abril 2009 se realizó una encuesta nacional de carácter transversal mediante cuestionario enviado a 35 centros, en los que se atiende a pacientes infantiles y adultos. La finalidad de la encuesta era establecer la prevalencia en ese momento recogiendo el histórico de pacientes que cada centro tuviera documentados.
ResultadosA través de los cuestionarios respondidos por 25 médicos de 16 centros, se han identificado 75 pacientes: 41 defectos de transulfuración (uno fallecido), 27 de remetilación (6 fallecidos) y 7 sin diagnóstico etiológico definitivo. La edad de diagnóstico muestra una amplia variación, en 18 casos había más de un hermano afectado. Las manifestaciones clínicas más graves inciden en el grupo de los pacientes afectados de trastornos de la remetilación. Destaca el alto porcentaje de déficit cognitivo, seguido de la patología de cristalino; casi la mitad de los pacientes presentan trastornos neurológicos, es elevada la afectación vascular en los adultos con deficiencia de CBS; las opciones terapéuticas más utilizadas han sido el ácido fólico, la hidroxicobalamina y la betaína.
ConclusionesA la vista de estos resultados, y en especial del escaso número de deficiencias de CBS detectadas, se concluye la necesidad de implantar el cribado neonatal para la homocistinuria clásica y asegurar la puesta en marcha del proceso diagnóstico oportuno en todos los pacientes de riesgo.
To determine the prevalence of homocystinuria in Spain and to establish the measures and mechanisms to ensure its prevention, diagnosis and treatment.
Material and methodsA national cross-sectional survey was conducted by means of a questionnaire sent to 35 hospitals in which children and adult patients are treated.
ResultsUsing the questionnaires submitted by 25 physicians from 16 centres, 75 patients were identified: 41 transsulphuration defects (one deceased), 27 remethylation (six deaths) and 7 without a syndromic diagnosis. The age at diagnosis varied widely, and 18 cases had more than one sibling affected. The more severe clinical manifestations involved the patients with remethylation defects. There was a high percentage of cognitive impairment, followed by lens diseases. Almost half of the patients had neurological disorders. There was increased vascular involvement in CBS-deficient adults. The therapeutic options most used were, folic acid, hydroxycobalamin and betaine.
ConclusionsIn view of these results and especially the small number of CBS deficiencies detected, we conclude that there is a need to introduce newborn screening for classical homocystinuria and ensure implementation of an appropriate diagnostic workup in all patients at risk.
Las homocistinurias abarcan un grupo de errores congénitos del metabolismo de los aminoácidos azufrados y de la cobalamina con una gran heterogeneidad etiológica y clínica; poseen sin embargo una serie de características comunes que justifican la revisión conjunta de su problemática sanitaria1–8.
Desde el punto de vista bioquímico, hay diferentes deficiencias enzimáticas que van a dar lugar a un aumento de homocisteína: trastornos de transulfuración de la homocistina, deficiencia de cistationina beta sintasa (CBS); trastornos de la remetilación de la homocisteína, deficiencia de metilen-tetrahidro-folato reductasa (MTHFR) y de la metionina sintasa (MS) y trastornos del metabolismo de la vitamina B12 que conducen a: a) la deficiencia exclusiva de la síntesis de metilcobalamina, cofactor de la MS, llamadas deficiencia de cobalamina E (CblE) y de cobalamina G (CblG), b) la deficiencia combinada de metilcobalamina, cofactor de la MS y de 5’deoxicobalamina, cofactor de la metil malonil CoA mutasa (MMCoAmut), denominadas deficiencias de transcobalamina II (TC II), de cobalamina C (CblC), de cobalamina D (CblD) y de cobalamina F (CblF) que cursan con homocistinuria y metilmalonico acidemia.
Regidas por una herencia de carácter autosómico recesivo, debutan habitualmente durante la infancia, pero pueden hacerlo en cualquier edad de la vida. Sus mecanismos fisiopatológicos tienen algunos componentes comunes9,10. Las manifestaciones clínicas son de carácter agudo y grave con elevadísimo riesgo de muerte en poco tiempo en las homocistinurias con metilmalónico acidemia; en las deficiencias de CBS, CblE, CbFl y T CII las manifestaciones clínicas pueden ser crónicas y progresivas, siendo en este grupo importante el diagnóstico precoz para prevenir el desarrollo de sintomatología. De forma general, todas ellas cursan fundamentalmente con afectación vascular, neurológica, oftalmológica y esquelética5–8.
Son entidades que se han considerado poco frecuentes (en torno a 1/200.000-300.000 recién nacidos), aunque se conoce la existencia de notables variaciones étnicas. Sin embargo, la progresiva instauración de los programas de cribado neonatal y la búsqueda sistemática de esta patología en los pacientes con manifestaciones clínicas compatibles (y no solamente en aquellos con un fenotipo «clásico» de deficiencia en CBS) ponen en evidencia creciente que su incidencia puede ser mucho más elevada de lo que se creía (alrededor de 1/20.000 recién nacidos) y que después de la hiperfenilalaninemia es probablemente la aminoacidopatía más frecuente5,6,9,11.
En algunas de estas deficiencias, CBS, TC II, CblE y CblF, se dispone en el momento actual de posibilidades terapéuticas muy efectivas cuando se aplican de forma precoz y antes de que se produzcan lesiones orgánicas irreversibles7.8.
ObjetivosElaboración de un registro nacional anonimizado de errores congénitos de los aminoácidos azufrados y del metabolismo de la cobalamina que producen hiperhomocistinemia, con el fin de recoger la problemática asistencial actual que esta patología genera en España, valorando su frecuencia, prevalencia, etiología, tratamientos utilizados y evolución general clínica. Ello debería permitir establecer las medidas asistenciales adecuadas para garantizar su prevención, diagnóstico y tratamiento adecuados.
Material y métodosSe ha realizado una encuesta de incidencia y prevalencia transversal mediante el envío de un sencillo cuestionario a todas las unidades de referencia para el diagnóstico y tratamiento de la enfermedades metabólicas hereditarias y a todos los centros y unidades en los que existía constancia de uso de betaína anhidra, por lo que se presupuso que se trataban pacientes con esta patología. Se pretendía recoger a fecha de abril-mayo 2009 el histórico de pacientes con errores congénitos del metabolismo que producen hiperhomocistinemia, documentados en los diferentes centros del estado español.
La encuesta incluía antecedentes familiares, datos de filiación, edad del diagnóstico, manifestaciones clínicas generales, perfil bioquímico en el momento del diagnóstico, estudios enzimáticos y moleculares realizados, diagnóstico definitivo, tratamiento recibido, supervivencia y seguimiento. Para facilitar su cumplimentación, el cuestionario se diseñó de forma sencilla para recoger datos esenciales. No se recogieron datos de dosificación de fármacos.
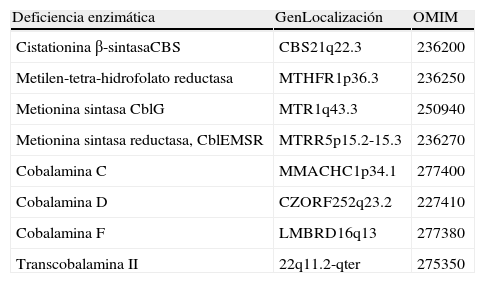
Los criterios de inclusión fueron: a) que el paciente estuviera afectado (con confirmación enzimática o molecular o diagnóstico bioquímico compatible) de alguno de los diagnósticos etiológicos recogidos en la tabla 1, y b) existencia del consentimiento del comité de ética del centro al que pertenecía para que sus datos pudieran ser incluidos en la encuesta.
Defectos metabólicos causantes de homocistinuria congénita
| Deficiencia enzimática | GenLocalización | OMIM |
| Cistationina β-sintasaCBS | CBS21q22.3 | 236200 |
| Metilen-tetra-hidrofolato reductasa | MTHFR1p36.3 | 236250 |
| Metionina sintasa CblG | MTR1q43.3 | 250940 |
| Metionina sintasa reductasa, CblEMSR | MTRR5p15.2-15.3 | 236270 |
| Cobalamina C | MMACHC1p34.1 | 277400 |
| Cobalamina D | CZORF252q23.2 | 227410 |
| Cobalamina F | LMBRD16q13 | 277380 |
| Transcobalamina II | 22q11.2-qter | 275350 |
Todos los cuestionarios recibidos se procesaron sin excluir ninguno a priori. Los datos se han recogido en una base de datos Microsoft Access y el tratamiento estadístico se ha realizado mediante SPSS 15.0.
ResultadosLa encuesta se envió a 35 centros y se obtuvieron respuestas adecuadas procedentes de 17 de ellos (50% de las encuestas enviadas) distribuidos homogéneamente por todo el territorio español.
Se han obtenido datos fiables, aunque no todos completos, de 75 pacientes. Cuarenta y seis de ellos (61,4%) hombres y 29 (38.6%) mujeres. De éstos son: 31 déficit CBS no sensible a B6 (40%), 4 déficit CBS sensible a B6 (5%), 8 déficit de MTHFR (11%), 3 (4%) déficit sistema metionina sintetasa (Cbl E, CblG), 22 (29%) déficit de CblC, Cbl D, Cbl E, y 8 casos sin filiar (11%).
Antecedentes familiaresExistían antecedentes de consanguinidad en 5 pacientes (6,6%). Dos de ellos eran deficiencias comprobadas y otro, una sospecha de MTHFR; un defecto de remetilación sin diagnóstico definitivo y una deficiencia de CBS. Se desconocía la existencia o no de consanguinidad en cuatro pacientes (5.3%). Dieciocho pacientes tenían otro hermano afecto, lo que supone que en el 24% de las familias se había repetido esta patología.
Tiempo de seguimientoSeis enfermos habían abandonado el control médico. Todos eran deficiencias de CBS, comprobadas mediante estudio enzimático o molecular en 4 casos y diagnosticadas por el perfil bioquímico en los otros dos.
EvoluciónHabían fallecido 6 pacientes. Cinco lactantes afectados de defectos de remetilación (2 cblC comprobados, otros 2 cblC sospechados, una deficiencia de MTHFR a los 14 años de edad y un déficit de CBS que debutó de un modo agudo con tromboembolismo cerebral masivo a los 10 años de vida).
Diagnóstico etiológicoAlgo más de la mitad de los pacientes (n = 41) habían sido diagnosticados de deficiencia de CBS. Treinta y cinco de ellos con estudio enzimático y/o molecular realizado y los 6 restantes con diagnóstico compatible por el perfil clínico y bioquímico. De todas las deficiencias de cistationina β-sintasa comprobadas sólo 4 habían sido catalogadas como sensibles a la piridoxina. En 20 familias con déficit de cistationina-β-sintasa se conoce el diagnóstico molecular. Quince son homocigotos para la mutación T191M, una para la p145L y una para la D444N. En una familia solo se identificó una mutación, la T353M. Dos pacientes eran dobles heterocigotos: T191M/I278S y T191M/D444N, respectivamente.
Las restantes 27 homocistinurias habían sido catalogadas de defectos de la remetilación. Diez de ellas con alta probabilidad diagnóstica por el perfil bioquímico, pero sin que se aporten resultados de otros exámenes complementarios. Cuatro pacientes se diagnosticaron de deficiencia de MTHFR, siendo uno de ellos portador de la mutación p.F435S en homocigosidad. Un caso con sospecha de transcobalamina II. Dos pacientes con sospecha de deficiencia de CblE y CblG, pendientes de resultados de complementación y moleculares. El subgrupo más numeroso de defectos de remetilación comprobados lo constituyen 9 pacientes diagnosticados de deficiencia de CblC, en tres de ellos debida a la mutación R91-fs.
En 7 pacientes los datos recibidos en la encuesta no permitieron una aproximación diagnóstica etiológica fiable.
Edad al diagnósticoLas edades al diagnóstico presentan una gran dispersión tal como se recoge en la figura 1. Llama la atención, no obstante, que el 6% de los pacientes fueron identificados en el periodo neonatal. Uno por cribado ampliado y los restantes por tener un hermano afectado de forma previa o por haber presentado una clínica aguda en esta época de la vida. Sin embargo, la mitad de los enfermos fueron diagnosticados por encima de los 10 años de vida, lo cual es, en los casos en los que un diagnóstico precoz permite poner terapéuticas eficaces, insuficiente para prevenir las consecuencias de la enfermedad.
Clínica
Las manifestaciones clínicas reportadas, consideradas en general, responden a la sintomatología clásica de las homocistinurias. Destaca el alto porcentaje de déficit cognitivo, seguido de la patología de cristalino; pero casi la mitad de los pacientes presentan trastornos neurológicos y la tercera parte patología vascular, osteoporosis-osteopenia y problemas de comportamiento (fig. 2).

Las manifestaciones clínicas más graves inciden en el grupo de los pacientes afectados de trastornos de la remetilación. Todos los fallecidos, menos uno, pertenecían a este grupo; 4 lo hicieron antes de los 3 meses de vida y el otro a los 14 meses. Todas las encuestas recogen retraso cognitivo, la mayoría de las ocasiones grave y que se acompaña con frecuencia de alteraciones neurorradiológicas como leucodistrofia, poliodistrofia o atrofia cortical, oftalmológicas (degeneración macular) y clínicas (hidrocefalia, hipotonía central, ausencia de adquisición de hitos de desarrollo, etc.). Son frecuentes asimismo las alteraciones hematológicas en forma de anemia macrocítica y pancitopenia fundamentalmente. Llama la atención la existencia de tres pacientes con déficit de CblC que fallecieron en las primeras semanas de vida con un grave cuadro de microangiopatía multiorgánica (fig. 3).
Tratamiento
Es necesario considerar que, con el fin de simplificar la cumplimentación de la encuesta, no se solicitó la cuantificación de los tratamientos ni el tiempo exacto de su aplicación.
En general, existe una gran uniformidad en el tratamiento según la etiología de estos pacientes, y las medidas terapéuticas utilizadas son desde el punto de vista cualitativo, las habitualmente recomendadas en la bibliografía para cada una de las patologías. Desde esta perspectiva es lógico que el ácido fólico, la hidroxicobalamina y la betaína sean las opciones terapéuticas más utilizadas. La piridoxina solo se ha utilizado en la mitad de los casos, seguramente en los déficit de cistationina b-sintasa, y el suplemento de cistina sólo se ha considerado necesario en el 22% de los pacientes (fig. 4). Existe un alto porcentaje de pacientes sometidos en algún momento a dieta limitada en metionina; no obstante, este dato no es analizable al no haber establecido el momento ni la duración del tratamiento.
Discusión
La prevalencia de los errores congénitos del metabolismo que cursan con homocistinuria en España, según los datos recogido en la encuesta nacional, sería de 1,6/1.000.000 de habitantes. Los 75 pacientes recogidos no reflejan probablemente la verdadera incidencia y prevalencia de los diferentes errores innatos del metabolismo que pueden presentar homocistinuria5,6,9,11. Es lógico suponer que un número significativo de deficiencias de CBS no han sido identificadas, ya que los 41 casos recogidos son una cifra inferior a la esperada según los datos de la bibliografía. Las causas podrían ser la gran heterogeneidad clínica en las deficiencias de transulfuración, y la situación del cribado neonatal para la homocistinuria clásica en España que sólo se realizaba en la comunidad autónoma de Galicia en el momento de la realización de la encuesta. Los errores del metabolismo de la CblC y la CblD suelen ser tan graves al debut que no sobreviven y en ocasiones fallecen sin establecerse el diagnóstico. Por el contrario, la importancia de las manifestaciones clínicas de los defectos de remetilación, mucho menos frecuentes en teoría, hace que el número de 27 pacientes diagnosticados de esta patología sea más coherente con las tasas de incidencia esperables.
La existencia de más de un hermano afectado en el 24% de los casos sugiere un diagnóstico tardío de la enfermedad en el primer hijo afectado o un fallo en el asesoramiento genético familiar.
La mutación génica responsable con más frecuencia de los defectos de transulfuración ha sido la T191M, situación característica de España y Portugal. Se trata de una mutación que en homocigosidad no responde a la vitamina B6, pero que no presenta otra relación definida con el fenotipo clínico. Las mutaciones probablemente más frecuentes en Europa, la I128T y la G307S, no han sido identificadas en este grupo de pacientes, en concordancia con los estudios moleculares previamente publicados en pacientes iberoamericanos. Entre los defectos de remetilación se ha identificado la mutación p.F435S en una deficiencia de MTHFR y la R91-fs en una deficiencia de CblC12–17.
Las manifestaciones clínicas de la deficiencia de CBS recogidas en la encuesta responden al fenotipo clínico característico de esta patología. Los defectos de transulfuración están precedidos por las deficiencias cognitivas y la patología ocular en los primeros años de la vida, mientras que las alteraciones de la mineralización ósea y los trastornos vasculares son prevalentes en los pacientes adultos. Estos perfiles clínicos son importantes para ayudar a definir las poblaciones de riesgo para esta patología18-20.
Los defectos de remetilación debutan en todos los casos durante las primeras semanas o meses de vida, con una importante patología neurológica y hematológica que compromete gravemente la supervivencia de los pacientes. De hecho, de los 7 pacientes fallecidos sólo uno era un defecto de CBS que debutó a los 10 años de edad con un cuadro masivo de tromboembolismo cerebral.
El tratamiento que los pacientes han recibido, desde el punto de vista de las opciones terapéuticas utilizadas, es el adecuado en todos los casos. Es posible que en algunos casos no se hayan podido evitar daños orgánicos ya irreversibles debido a la gran dispersión de las edades en el momento del diagnóstico.
La encuesta realizada tiene unos puntos fuertes que son la cobertura nacional conseguida, la recogida de datos de niños y adultos y el hecho de ser la primera vez que se han obtenido datos epidemiológicos y clínicos de esta patología en España. Como puntos débiles destacan la falta de datos acerca de las dosis y el momento de aplicación de los distintos tratamientos realizados y la presencia de un 9,3% de pacientes cuyo diagnóstico etiológico no han podido ser correctamente etiquetado a través de los datos reseñados en el formulario de la encuesta.
El cribado neonatal de los errores congénitos del metabolismo de la homocisteína, metionina y cobalamina se apoya en las concentraciones anómalas de la metionina y la propionilcarnitina. Estos analitos, no obstante, no son específicos y dan lugar a frecuentes falsos positivos. La homocisteína total (tHCY), el ácido metilmalónico (MMA) y el ácido metilcítrico (MCA) son marcadores más específicos, pero en el momento actual no pueden ser detectados con los métodos utilizados en el cribado expandido. Se están desarrollando nuevas metodologías para mejorara esta situación20 mediante la determinación de tHCY, MMA y MCA en las muestras de sangre seca mediante cromatografía líquida-espectrometría de masas en tándem (LC-MS/MS)21.
ConclusionesEn resumen, de acuerdo con los datos recogidos en la encuesta nacional, la prevalencia de los defectos de la transulfuración en España presenta una frecuencia menor a la descrita en la bibliografía.
El momento del diagnóstico de los pacientes presenta una amplia variabilidad etaria.
Destaca la frecuencia de la patología vascular como la manifestación clínica inicial más importante en el adulto. La difusión y la divulgación de claves para el diagnóstico en atención primaria y especializada de esta patología permitirán la identificación de pacientes, niños o adultos, de riesgo para esta patología.
Es importante la implantación del cribado neonatal de la homocistinuria clásica por defecto de CBS y de los defectos de la remetilación en todo el territorio nacional con el fin de asegurar su tratamiento precoz.
FinanciaciónEl trabajo ha sido realizado con la ayuda de una beca de la Asociación Española para el Estudio de los Errores Congénitos del Metabolismo (AECOM).
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Dra. Consuelo Fernández-Miranda, Dr. Josep M. Aragonés, Dr. Jose Ignacio Martínez-Berniz, Dra. Verónica González Álvarez, Dra. Belén Pérez Dueña, Dr. Luis Aldámiz-Echevarría, Dra. María T Forga, Dra. Olga Alonso Luengo, Dr. Eduardo Martínez García-Boné, Dra. Mónica Ruiz, Dra. María Bueno Delgado, Dra. Mireya Del Toro y Dra. Rosa Lama More.
