
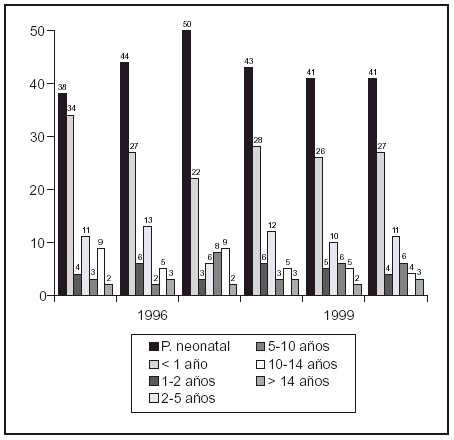
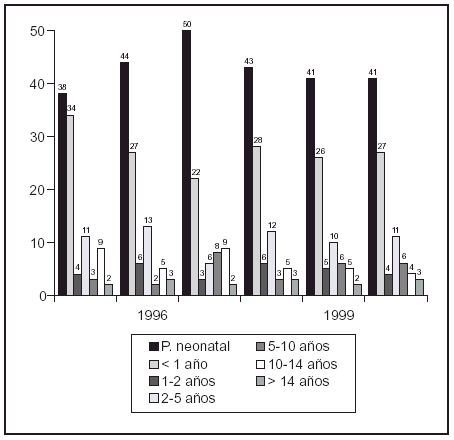
P578
ABDOMEN AGUDO: FORMA POCO FRECUENTE DE PRESENTACIÓN DE LINFOMA
P.M. Santos Rodríguez, J.L. Otero Díez, I. Riaño Galán, B. Lastra Areces y P. Fernández González
Hospital Carmen y Severo Ochoa, Cangas del Narcea.
Introducción: El linfoma es la tercera forma de cáncer más frecuente en niños. Un 31% de los linfomas no hodgkinianos se presentan primariamente en abdomen, y suelen localizarse en la región ileocecal.
Caso clínico: Varón de 8 años, que acude a urgencias por dolor abdominal tipo cólico de diez días de evolución, al que en los dos últimos días se le había asociado diarrea y febrícula. Proceso catarral de vías altas, los días previos. Antecedentes familiares y personales sin interés. Exploración física: buen estado general, afebril, no lesiones cutáneas ni adenopatías, destacando solamente el abdomen moderadamente distendido y timpanizado, doloroso a la palpación profunda en hipogastrio y FII, donde presenta sensación de masa dolorosa. No hay defensa ni contractura abdominal. En el tacto rectal se aprecia masa anterior extraluminal, dura y no dolorosa que estrecha la ampolla rectal. Exámenes complementarios: Hemograma discreta anemia microcítica e hipocrómica; Bioquímica normal. Radiografía de tórax: sin hallazgos. Radiografía de abdomen: Distensión en asas de intestino delgado. Ecografía Abdominal: Liquido libre intraperitoneal con distribución predominante en flanco dcho, ciego edematoso y rodeado de liquido. Ante la sospecha de absceso pélvico de posible origen apendicular, se realiza cirugía urgente. Tras laparotomía media infraumbilical, aparece gran cantidad de liquido ascítico claro, y gran nº de tumoraciones fundamentalmente retroperitoneales, que infiltran los mesos e improntan en intestino y peritoneo parietal. Biopsia intraoperatoria: tumor maligno de células redondas. TAC torácico y abdomino-pélvico postoperatorio: derrame pleural bilateral más intenso en lado dcho, sin adenopatías mediastínicas. Discreta lobulación anterior en lóbulo hepático izquierdo, con parénquima hepático restante normal. Con la sospecha de carcinomatosis por tumor maligno de células redondas, es trasladado al centro de referencia. Sin confirmación anatomopatológica definitiva, se diagnostica como Linfoma Linfoblástico "Burkitt like", con participación abdominal y derrame pleural bilateral, sin afectación de lcr ni médula ósea (estadio III de Murphi). Dada la progresión del tumor se inicia tratamiento con esteroides durante 72 horas, y posteriormente protocolo PATTE. Se constata la remisión completa.
Comentario: Los tumores malignos son causa poco frecuente de abdomen agudo en la infancia, y pueden simular otro tipo de patologías más habituales como apendicitis, invaginación o adenitis mesentérica.
P579
SÍNDROME DEL NEVUS EPIDÉRMICO ASOCIADO A RABDOMIOSARCOMA DE PRÓSTATA: CASO CLÍNICO
P. Tejado Merino, J.J. Casañ Plaza, G. García Matas, F. Vela Casas, J. Sánchez Calero y J. González-Hachero
Hospital Universitario Virgen Macarena, Sevilla.
Introducción: El síndrome del nevus epidérmico (SNE) es un cuadro cutáneo poco frecuente caracterizado por la presencia de uno o varios tipos de nevus epidérmicos (Nevus epidérmicos verrugosos, nevus sebáceos, comedonevus) de localización múltiple, pudiendo asociar otras manifestaciones cutáneas, así como alteraciones oculares, óseas y del sistema nervioso. En ocasiones, se asocia a neoplasias malignas sistémicas en niños y adultos jóvenes.
Caso clínico: Varón de 4 años que consulta por disuria con retención urinaria de varios días de evolución. Antecedentes personales: Controlado desde el nacimiento por la presencia de múltiples nevus sebáceos. Exploración clínica: Buen estado general, presentando múltiples nevus sebáceos en cuero cabelludo, cara y espalda, y múltiples nevus melanocíticos de pequeño tamaño en hemicuerpo superior izquierdo que, en el tronco, asientan sobre una mácula café con leche; en conjuntiva se observan 2 neoformaciones hipercrómicas de 1 mm de diámetro. A nivel suprapúbico se palpa una masa de consistencia dura y bien delimitada de 5 cm de diámetro, no dolorosa. El resto de la exploración es normal. Pruebas complementarias: Hemograma, bioquímica y coagulación normales; VSG: 31 mm en la 1ª hora; LDH: 736 U/L; sedimento urinario: 10-15 hematíes por campo; ecografía abdominal: tumoración sólida bien definida infravesical; RNM: masa pélvica entre vejiga y recto de probable origen prostático que no infiltra órganos adyacentes; estudio de médula ósea, gammagrafía ósea, TAC de tórax y marcadores tumorales normales. Biopsia de cilindro tumoral: Rabdomiosarcoma embrionario de origen prostático. Con este diagnóstico se inició tratamiento según el protocolo MMT-95 modificado para rabdomiosarcoma de la Sociedad Española de Oncología Pediátrica.
Conclusión: Presentamos un caso de SNE asociado a una neoplasia maligna sistémica en un niño. Aunque no hemos encontrado ninguna referencia respecto a la asociación entre SNE y rabdomiosarcoma de próstata, la asociación de este síndrome a neoplasias malignas en niños y jóvenes, en numerosas ocasiones de localización urogenital, nos hace pensar que esta asociación se trata de algo más que de una coincidencia.
P580
TUMORES GERMINALES: NUESTRA CASUÍSTICA
M.E. Rojas Gracia, A. Herrero Hernández, M.J. Ortega-Acosta, T. Acha García, A. Jurado Ortiz y T.J. Martínez Arán
Hospital Materno Infantil, Málaga.
Material y método: Estudio retrospectivo de todos los casos de niños diagnosticados y tratados de tumores germinales en el servicio de oncología pediátrica desde el año 1981 hasta enero del 2002.
Resultados: Total de 48 casos, con edad media de 4,7 años. Menores de 4 meses el 20,8%, siendo el 100% teratomas, predominantemente sacrocoxígeo. Desde los 4 meses a los 5 años el 37,5%, siendo la localización más frecuente el testículo, y una histología predominante de teratoma o tumor del seno endodérmico. Mayores de 5 años el 41,6%, siendo la localización más frecuente el ovario, y una histología predominante de teratoma. Hay un predominio femenino (64,6%), con relación varón/mujer1/1,8. Localización: ovario 43,7%, testículo 18,7%, sacrocoxígeo 18,7%, abdomen 6,2%, cerebral 6,2%, mediastino 4,1%, cabeza y cuello 2%. La forma de presentación clínica más frecuente de la localización ovárica fue el dolor abdominal, seguido de masa. El 100% de los tumores testiculares y sacrocoxígeos se presentaron como masa. Anatomía patológica: teratomas 66,6%, tumor del seno endodérmico 14,5%, tumor de células germinales mixto 12,5%, germinoma 4,2%, coriocarcinoma 2,1%. Estadiaje: I 72,9%, II 4,2%, III 16,6%, IV 6,3%. Tratamiento: Cirugía en 47/48 casos: resección completa (42) 89,3%, parcial (3) 6,3%, biopsia (2) 4,2%. Quimioterapia en 14/48, 12 en el tratamiento inicial, 5 en el tratamiento de recidivas. Radioterapia en un caso (germinoma cerebral). El tiempo medio de seguimiento es de 7,3 años. Se detectaron 7 recidivas, 71,4% en tumores extragonadales. Se produjeron 3 fallecimientos ( teratoma cerebral inmaduro E III, coriocarcinoma E IV, tumor mixto sacrocoxígeo E III). Se perdieron en su seguimiento 2 pacientes. La supervivencia global es del 93,4% (43/46) y la supervivencia libre de eventos del 89,13% (41/46).
Conclusiones: Los tumores de células germinales más frecuentes en nuestra serie han sido los teratomas maduros, de localización predominantemente ovárico, estadio I. En un 90% de los casos se realizó resección quirúrgica completa. La supervivencia libre de eventos es del 88,8% con un tiempo medio de seguimiento de 7,3 años. La localización extragonadal determina un peor pronóstico.
P581
SÍNDROME DE POLAND Y CARCINOMA NASO-FARÍNGEO
A. Remesal Escalero, D. Fernández-Álvarez, L. San Feliciano Martín, M. Muriel, J.A. Martín García y G. Mateos Pérez
Hospital Clínico Universitario, Salamanca.
El síndrome de Poland es una anomalía congénita que presenta de forma variable aplasia de la cabeza del esternón, del músculo pectoral mayor, hipoplasia de extremidad superior homolateral y aplasia del II, III, IV y V dedo. Aunque es un proceso relativamente conocido, pocas publicaciones lo relacionan con patología tumoral.
Presentamos un caso con aplasia del pectoral, hipoplasia mamilar ipsilateral y carcinoma nasofaríngeo.
Caso clínico: Se trata de un paciente de 13 años con historia de mes y medio de evolución consistente en otalgia, adenopatía laterocervical derecha, hipoacusia; odinofagia y fiebre. En la exploración además se objetiva tórax asimétrico con ausencia de pectoral mayor derecho e hipoplasia mamila derecha; protrusión mitad derecha del velo del paladar y pared lateral derecha de orofaringe. Por persistencia de adenopatía a pesar del tratamiento antibiótico se realiza PAAF de la misma que informan de carcinoma indiferenciado. Se realiza estudio de imagen mediante TAC y RMN cervico-craneal y torácico: masa en cavum que impronta y distorsiona su pared con obstrucción parcial de la vía aérea que se extiende hacia base de cráneo; adenopatías bilaterales en cadenas prevascular y cervical posterior. Se detectan Ac para VEB +, IgG e IgM. Otros marcadores tumorales negativos. Recibe quimioterapia y radioterapia con remisión completa a su finalización, actualmente al año del diagnóstico está libre de enfermedad.
Comentarios: Destacamos el interés de este síndrome y la asociación con procesos malignos hematológicos o tumores sólidos descritos recientemente en la literatura. Se sugiere que la mayor incidencia de enfermedades malignas forma parte de la anomalía genética subyacente. Carcinoma nasofaríngeo es raro en niños y se presentan con metástasis ganglionares frecuentes (60-90%) mostrando supervivencia libre de evento a los 5 años de 37-60% según estadio. Carcinoma nasofaríngeo se asocia a infección por VEB y síndrome de Poland, circunstancias poco frecuentes que se dan en nuestro paciente.
P582
ESCLEROSIS TUBEROSA Y CARCINOMA RENAL EN LA INFANCIA
J.L. Gómez Llorente, M.A. Vázquez López, F. Lendínez Molinos, M. Leyva Carmona, F.J. Aguirre Rodríguez, M.R. Jiménez Liria, M.A. Llamas Guisado, J. Momblan de Cabo y J. López Muñoz
Hospital Torrecárdenas, Almería.
Introducción: La esclerosis tuberosa (ET) es un trastorno neurocutáneo con afectación multisistémica y trasmitido genéticamente. La afectación renal aparece en más del 50% de los casos, siendo característicos los angiomiolipomas y quistes renales. El carcinoma renal (CR) es raro en la infancia y su presencia en niños con ET es igualmente excepcional. Presentamos el caso de una niña con ET y CR y se discute la problemática diagnóstica y el adecuado proceder terapéutico.
Caso clínico: Paciente de 10 años portadora de esclerosis tuberosa que ingresó para estudio de tumoración renal izquierda detectada en control ecográfico realizado para diagnóstico de patología apendicular en el curso de episodio abdominal agudo. La paciente fue diagnosticada de ET a la edad de 5 meses y en la actualidad presenta retraso psicomotor y estigmas en piel y SNC propios de la enfermedad de base. El estudio radiológico abdominal realizado durante el proceso apendicular agudo (ecografía y TAC) mostró la existencia de una lesión sólida de 2x3 cm localizada en polo superior de riñón izquierdo. El estudio de extensión que incluyó TAC torácico y gammagrafía ósea no mostró anormalidades. Tras laparotomía se realiza nefrectomía parcial izquierda con exéresis completa de la tumoración, siendo el diagnóstico anatomopatológico compatible con Carcinoma Renal (Estadio 1). El estudio radiológico (RNM abdominal) realizado a los 3 meses de la cirugía ha evidenciado una lesión de 1 cm localizada en polo superior de riñón contralateral, con características de señal propias de angiomiolipoma que requerirá seguimiento estrecho, valorando su crecimiento y la necesidad de estudio anatomopatológico.
Comentarios: Importancia de realizar un screening radiológico abdominal seriado en niños con ET para la detección precoz de lesiones renales. Radiológicamente el CR puede presentar problemas de D/D con otras lesiones benignas propias de la ET como son los angiomiolipomas. La actitud terapéutica quirúrgica recomendada para el CR en la ET debe ser conservadora dada la posibilidad de presentar lesiones múltiples y/o bilaterales que conlleven el sacrificio de mayor cantidad de parenquima renal.
P583
CRISIS HIPERTENSIVA COMO CLÍNICA INICIAL DEL NEUROBLASTOMA
C. Muñoz Román, G. Ramírez Villar, C. Márquez Vega, C. Montero Valladares, G. Calderón López, M.L. Anguita Quesada, E. Quiroga Cantero, A. Vilaplana López, A. Valladares Otero y A.M. Álvarez Silván
Hospital Universitario Virgen del Rocío, Sevilla.
Introducción: El neuroblastoma es en nuestro medio el tumor sólido extracraneal más frecuente en la infancia. Tiene un origen neural y puede desarrollarse en cualquier localización del sistema nervioso simpático, siendo la más frecuente a nivel abdominal (glándula suprarrenal, ganglios paraespinales). Pocos tumores tienen una sintomatología tan polimorfa como el neuroblastoma debido a la variedad de localizaciones, a su diseminación precoz y a la posibilidad de producir síndrome paraneoplásico.
Presentamos el caso de una niña de 14 meses a la que durante el estudio por hipertensión arterial se le detecta masa suprarrenal derecha confirmándose el diagnóstico de neuroblastoma tras laparotomía.
Caso clínico: Niña de 14 meses que ingresa para estudio de hipertensión arterial detectada en el curso de una gastroenteritis.Durante el ingreso sufre crisis hipertensiva (TAS 200 mmHg) con hemiparesia izquierda y desviación de la cabeza a la derecha, precisando ingreso en UCI donde se estabiliza tras instaurar tratamiento anticoagulante y antihipertensivo. Se realiza RMN cerebral presentando lesiones de isquemia en el territorio de la arteria cerebral media izquierda. En TAC abdominal se descubre masa suprarrenal derecha de unos 3 cm con desplazamiento caudal del riñón derecho y en la arteriografía renal se observa una obstrucción completa del flujo de la arteria renal derecha, obteniéndose imágenes de vascularización y renograma tras avanzar el catéter más allá del punto obstructivo.
La sospecha de neuroblastoma se confirma tras extirpación de la tumoración y estudio histológico de la misma. Al mismo tiempo se realiza estudio de hipercoagulabilidad ante la sospecha de trombosis de la arteria renal como causa de la hipertensión (estudio de coagulación, proteína C, proteína S, antitrombina III, anticuerpos antifosfolípido) que se encontraban dentro de los límites de la normalidad.
Comentarios: La hipertensión arterial es una de las manifestaciones clínicas del neuroblastoma por elevación de catecolaminas. En nuestro caso, la hipertensión arterial y la crisis hipertensiva detectada tiene un origen renovascular que inicialmente obligó a realizar un diagnóstico diferencial entre obstrucción arterial intrínseca por trombosis y una posible situación de hipercoagulabilidad, malformación renal asociada a neuroblastoma o una obstrucción arterial extrínseca por elongación o compresión de la arteria renal en relación con la tumoración suprarrenal.
P584
HEPATOMEGALIA ASINTOMÁTICA DE ETIOLOGÍA INFRECUENTE
R.A. Montesdeoca Melián, R. Perera Soler, J.L. Aparicio Sánchez, M.T. Herráiz Culebras y R. López Almaraz
Hospital Universitario de Canarias, La Laguna.
Introducción: La hepatomegalia constituye un hallazgo exploratorio relativamente frecuente en la edad pediátrica, representando la expresión clínica de una patología casi siempre benigna. Si bien las neoformaciones malignas hepáticas en niños son muy infrecuentes en nuestro medio, hay que tenerlas en cuenta ante la palpación de una hepatomegalia asintomática.
Caso clínico: Paciente varón de 16 meses de edad sin antecedentes personales de interés que presenta fiebre (hasta 39 ºC) y pérdida de apetito de tres días de evolución. En la exploración física destaca la presencia de una hepatomegalia dura, de tres centímetros por debajo del reborde costal, no dolorosa sin otros hallazgos de interés.
En los exámenes complementarios practicados se observa: hemograma y perfil bioquímico normal con LDH de 3.292 U/L y alfa-fetoproteína de 39.576 ng/ml. En la ecografía abdominal presenta una masa en lóbulo hepático derecho de 7 x 6,4 cm que desplaza la vesícula biliar, con áreas hipoecoicas (compatibles con necrosis) y sin compromiso vascular. TAC toraco-abdomino-pélvico: masa heterogénea con áreas hipodensas en su interior, en probable relación con necrosis y calcificaciones puntiformes sin que se observan adenopatías. RMN hepática con contraste en la que se observa voluminosa tumoración hepática, esférica, bien delimitada respecto al parénquima de 6 x 6 cm, sin signos de trombosis portal. Gammagrafía ósea negativa. Con los datos obtenidos se llega al diagnóstico de Hepatoblastoma (estadio IIa prequirúrgico), instaurándose quimioterapia según protocolo SIOPEL-1.
Conclusión: Ante toda hepatomegalia asintomática se debe incluir en el diagnóstico diferencial a los procesos tumorales aún siendo infrecuentes en la edad pediátrica, y realizar de entrada una ecografía abdominal.
P585
HISTIOCITOSIS DE CÉLULAS DE LANGERHANS: CASUÍSTICA EN NUESTRO CENTRO
M.I. Llull Ferretjans, R. Amo Rodríguez, N. Nieto del Rincón, M. Guibelalde del Castillo, J.M. Román Piñana, M. Herrera Savall y J. Mulet Ferragut
Hospital Son Dureta, Palma de Mallorca.
Antecedentes: La histiocitosis de células de Langerhans (HCL) es una enfermedad producida por infiltración en uno o más órganos de células de Langerhans patológicas.
Objetivos:1) Revisar la clínica al diagnóstico de nuestros pacientes. 2) Estudiar su tratamiento y evolución. 3) Valorar el enfoque diagnóstico y terapéutico realizado por las distintas especialidades.
Métodos: Revisión de las historias clínicas de los pacientes pediátricos (hasta 14 años), diagnosticados de HCL entre enero 1988 y diciembre 2001.
Resultados: En los 14 años revisados se diagnosticaron 13 casos de HCL, todos varones. Su edad media fue de 3 años (rango de 10 días-14 años).Cuatro de ellos fueron menores de 2 años. La clínica al diagnóstico fue: asintomáticos en 2, tumoración partes blandas en 5, dolor en 3, disfagia en 1, disnea y exantema en 1, otorrea y exantema en 1. La enfermedad fue localizada en 9 (ósea en 8, adenopatías en 1) y multisistémica en 4 (1º: 2 huesos; 2º: múltiples lesiones óseas y adenopatías; 3º: piel y pulmón; 4º: piel y oido). La localización ósea más frecuente fue el cráneo (6 casos). La enfermedad localizada se trató con cirugía en 3 casos, infiltración de corticoides en 2 y tratamiento conservador en 4.Los pacientes con enfermedad multisistémica (EM), recibieron quimioterapia (corticoides, Vinblastina, VP, ciclofosfamida, metotrexate). Tras el tratamiento inicial alcanzaron la remisión 12 pacientes, de los que 5 recidivaron (2 de ellos con EM al diagnóstico). Éstos se trataron con quimioterapia 3, infiltración de corticoides 1, cirugía 1 y no se trataron 2, consiguiendo todos remisión completa. El paciente refractario al tratamiento inicial, recibió quimioterapia en las recaídas y falleció por progresión (afectación multiorgánica). De los 4 niños menores de 2 años, 3 han presentado recidivas y 2 afectación EM con disfunción orgánica. Las diferentes especialidades pediátricas realizaron diferentes pruebas diagnósticas, pero el tratamiento empleado fue similar.
Conclusiones:1) Predominio sexo masculino 2) El hueso es el órgano más afectado 3) Las formas localizadas presentan evolución favorable 4) Los pacientes menores de 2 años han presentado formas más severas y con más recidivas. 5) Un protocolo común facilitaría el manejo y seguimiento de los pacientes.
P586
MIOSITIS OSIFICANTE DE PRESENTACIÓN ATÍPICA
A. González Calvar, N. Nieto del Rincón, M. Guibelalde del Castillo, M. Herrera Savall y J.I. Bregante Ucedo
Hospital Son Dureta, Palma de Mallorca.
Presentación y objetivos: La miositis osificante es una entidad clínica poco frecuente que hay que tener presente dentro del diagnóstico diferencial de las masas de partes blandas. Presentamos como ejemplo el siguiente caso clínico.
Caso clínico: Niña de 12 años de edad, que consulta por masa en región pectoral derecha, dolorosa a la palpación y a la movilización del hombro ipsilateral, de 3 semanas de evolución, que ha aumentado de tamaño la última semana. No refiere traumatismos previos. Jugadora de baloncesto. No asocia síndrome constitucional, dificultad respiratoria, ni alteraciones digestivas. No antecedentes patológicos de interés. Antecedentes familiares: hermano de 8 años intervenido por atresia esofágica, abuelo materno fallecido por carcinoma de próstata.
Exploración física: Buen estado general y nutricional, no lesiones cutáneas ni adenopatías. Masa de 3*4 cm de diámetro en región pectoral derecha, de consistencia dura, mal delimitada del tejido circundante, fija a planos profundos y dolorosa a la palpación, con piel que recubre la lesión sin signos inflamatorios. Resto de exploración por aparatos y sistemas normal.
Pruebas complementarias: Hemograma, bioquímica y coagulación normales. Radiografía de tórax normal. Resonancia torácica: masa sólida dentro del pectoral mayor derecho, bien delimitada. Tomografía torácica: masa dentro del pectoral mayor derecho, con calcificación en la periferia y agrandamiento difuso del músculo. Ecografía abdomen: normal. Gammagrafía ósea: normal.
Actitud diagnóstica y terapéutica: Se decidió realizar resección de la lesión con márgenes amplios y estudio anatomopatológico de la misma, que demostró la existencia de un componente fusocelular de apariencia fibroblástica-miofibroblástica, zonas de material osteoide en el centro y metaplasia ósea madura en la periferia, compatible con miositis osificante.
Comentarios:1) La búsqueda de antecedentes traumáticos, puede ayudar a sospechar esta entidad, dentro de otras entidades de masas de partes blandas. 2) La demostración de la lesión radiológica típica, con calcificación periférica, fue posible gracias a la tomografía, mientras que la resonancia no ayudó para ello.
P587
MEDULOBLASTOMA EN LA EDAD PEDIÁTRICA: PRESENTACIÓN DE UNA CASUÍSTICA
M.O. Blanco Barca, M. López Rivas, A. Urisarri-Ruiz de Cortázar, A. Alonso Martín, M. Gelabert González, M.C. Porto Vázquez y J.M. Couselo Sánchez
Hospital Clínico Universitario - Complejo Hospitalario Universitario de Santiago de Compostela, Santiago de Compostela.
Objetivo: Evaluar aspectos epidemiológicos, clínicos, resultados terapéuticos y complicaciones de niños afectos de meduloblastoma (MB).
Metodología y resultados: Estudio retrospectivo en pacientes diagnosticados y tratados por MB en un hospital desde 1977 hasta la actualidad:

Conclusiones:a) La supervivencia de la muestra estudiada a los 9 años es del 46,7%, superponible a lo referido por la bibliografía. b) Los pacientes diagnosticados entre los años 1991-2002 tienen una supervivencia mayor que los diagnosticados entre 1977-90 (57,5 vs 37,1%). c) Un porcentaje alto de niños presentaron complicaciones agudas post-cirugía (53%) y secuelas a largo plazo (46,6%).
P588
INFILTRACIÓN PAROTÍDEA COMO PRIMERA MANIFESTACIÓN DE LEUCEMIA LINFOBLÁSTICA AGUDA TIPO T
G. Domínguez Ortega, A. Pérez Martínez, M.C. Martínez Martín y L. Madero López
Hospital del Niño Jesús, Madrid.
Introducción: La LLA es la neoplasia más común en la infancia, representando una cuarta parte de los cánceres en edad pediátrica. El pico de incidencia es entre los 3 a 5 años. La LLA tipo T supone el 13-15% de todas las LLA. Presentamos un caso atípico de LLA tipo T cuya manifestación inicial fue infiltración de glándulas parótidas.
Caso clínico: Niño de 3 años y medio que consulta por inflamación parotídea de 10 días de evolución. Diagnosticado de parotiditis inicia tratamiento con antiinflamatorios, con aumento progresivo de la consistencia y tamaño parotídeo. En la exploración se objetiva hipertrofia parotídea con consistencia leñosa, hepatomegalia y renomegalia. Se realiza análisis de sangre: Hb: 10,7 g/dl; leucocitos: 243.000/microlitro (96% blastos); plaquetas 103.000/mm3. Bioquímica: normal salvo LDH: 7584 /L, ácido úrico: 8,8 mg/dl. Se inicia tratamiento profiláctico de lisis tumoral. Serologías para hepatitis viral, varicela, VIH y prueba de la tuberculina: negativas. Aspirado de médula ósea: medulograma: LLA tipo L2, citoquímica: peroxidasa negativa, citrometría de flujo: CD1a 27%, inmunofenotipo: estirpe T, subtipo pre-T-intermedia, citogenética: 46 XY, traslocación t(7:14) (p13;q11) y estudios moleculares: reordenamiento TcRy positivo. Rx tórax: sin alteraciones. Punción lumbar: negativa para malignidad. Hemocultivo, urocultivo: estériles. Ecografía abdominal: riñones aumentados de tamaño de forma homogénea, hígado y bazo normales. Con el diagnóstico de LLA tipo T, se inicia tratamiento según el protocolo BFM 95. Al día +8 presenta 178 blastos en sangre periférica. Alcanza remisión completa medular en el día +33. Actualmente se encuentra en rama de terapia MR-2 del protocolo MCA del BFM 95.
Comentarios: La parotiditis es una patología banal pero que puede enmascarar cuadros más graves que precisen tratamientos agresivos y precoces. El diagnóstico y el tratamiento precoces mejoran el pronóstico de la LLA tipo T, por lo que debe incluirse en el diagnóstico diferencial de la hipertrofia parotídea.
P589
ASTROCITOMA DE BAJO GRADO DE MALIGNIDAD: CASUÍSTICA DE 20 AÑOS
A. Urisarri-Ruiz de Cortázar, M. López Rivas, M.O. Blanco Barca, A. Álvarez Moreno, L. Cascallar Canedo, J. Fernández Villa y J.M. Couselo Sánchez
Hospital Clínico Universitario - Complejo Hospitalario Universitario de Santiago de Compostela, Santiago de Compostela.
Objetivo: Evaluar epidemiología, clínica, tratamiento, evolución y complicaciones en niños diagnosticados de astrocitomas de bajo grado de malignidad (ABGM).
Metodología: Estudio retrospectivo de niños con ABGM en un centro hospitalario desde el año 1982 hasta 2002, valorando: edad, sexo, clínico de presentación, localización, tratamiento realizado, evolución y complicaciones.
Resultados:

Conclusiones:1) La supervivencia global de la serie estudiada (83,3%) y la supervivencia sin progresión de enfermedad (75%) es similar a la descrita en la bibliografía. 2) El grado de resección tumoral en esta serie ha sido el factor más determinante en la supervivencia. 3) Una tercera parte de los pacientes presentaron complicaciones posquirúrgicas y la mitad secuelas a largo plazo.
