









Next Generation Sequencing (NGS) encompasses a range of technologies that have transformed genomic research since the 2000s. By allowing the sequencing of large DNA fragments at a significantly lower cost than Sanger sequencing, NGS has become an indispensable tool in molecular laboratories, particularly in the field of molecular genetics. Its high efficiency and speed make it a first-line technique in genetic analysis.
A crucial step in achieving a diagnosis is bioinformatics analysis. Short-read sequencing technology generates raw data that must be processed to extract meaningful and interpretable information. This process enables the identification of causal links between genetic findings and phenotypic traits. Clinical bioinformatics specialists carry out this analysis using specialized tools and pipelines, which take into account the specific characteristics of the sequencing platforms, protocols and the particular diseases under study.
The quality review is an essential complement to the pipeline analysis. Its primary objective is to assess which samples are suitable for diagnosis and, in cases where results are negative, to identify the reasons, whether they are related to the incidence or other factors. Additionally, the quality review offers insight into the overall effectiveness of the experimental procedures.
Despite its many advantages, NGS still faces several challenges, including the need for more efficient technologies, enhanced regulatory frameworks and improved training of medical staff.
La secuenciación de nueva generación (NGS) abarca una gama de tecnologías que han transformado la investigación genómica desde los años 2000. Al permitir la secuenciación de grandes fragmentos de ADN a un costo significativamente menor que la secuenciación de Sanger, la NGS se ha convertido en una herramienta indispensable en los laboratorios moleculares, particularmente en el campo de la genética molecular. Su alta eficiencia y rapidez la convierten en una tecnología de primera línea en el análisis genético.
Un paso crucial para lograr un diagnóstico es el análisis bioinformático. La tecnología de secuenciación de lectura corta genera datos en bruto que deben ser procesados para extraer información significativa e interpretable. Este proceso permite la identificación de vínculos causales entre los hallazgos genéticos y los rasgos fenotípicos. Expertos en Bioinformática Clínica realizan este análisis utilizando herramientas especializadas y pipelines, que tienen en cuenta las características específicas de las plataformas de secuenciación, los protocolos y las enfermedades particulares que se están estudiando.
La revisión de calidad es un complemento esencial del análisis de las pipelines. Su objetivo principal es evaluar qué muestras son adecuadas para el diagnóstico y, en casos donde los resultados son negativos, identificar las razones, ya sean relacionadas con la incidencia u otros factores. Además, la revisión de calidad ofrece información sobre la efectividad general de los procedimientos experimentales.
A pesar de sus muchas ventajas, la NGS aún enfrenta varios desafíos, como la necesidad de tecnologías más eficientes, marcos regulatorios mejorados y una mejor capacitación para el personal médico.
Next-generation sequencing (NGS) is an umbrella term encompassing a range of DNA sequencing technologies1 that have revolutionized genomic research since the 2000s by enabling interrogation of large genomic regions at lower cost and in less time than Sanger sequencing. It has become an essential tool in molecular laboratories and is currently considered a first-line diagnostic method on account of its high throughput and rapid turnaround.
Today, NGS is used in multiple clinical settings, although its implementation is still uneven. Beyond molecular genetics, NGS is applied in oncology, microbiology, immunology, hematology and public health.
NGS platforms are divided into two broad categories: short-read platforms, which typically generate reads of approximately 100–300 base pairs,2 and long-read platforms3 (third-generation sequencing), which can produce reads of up to 100 000 base pairs, such as those from Pacific Biosciences and Oxford Nanopore Technologies. Illumina short-read platforms are the most widely used in clinical practice, and their workflows are widely regarded as a de facto standard.
In molecular genetics, short-read sequencing data must be processed to convert raw reads into useful, interpretable information that can be utilized to identify causal relationships between genetic findings and phenotypes and to guide management when clinically actionable findings are identified. Data analysis should be performed by specialists in clinical bioinformatics using dedicated tools that account for platform-specific characteristics, sequencing protocols and the particular diseases under study, typically implemented as analysis platforms or bioinformatics pipelines.
In recent years, multiple consortia have contributed valuable information and resources that have enabled a more thorough characterization of genomic variation in both healthy4 and diseased individuals,5 thereby improving the interpretability of NGS findings.
However, the application of high-throughput sequencing in diagnosis still faces multiple technical and methodological challenges,6,7 including the need for more efficient sequencing platforms, optimized bioinformatics algorithms, stronger regulatory and quality-control frameworks and improved training for health care professionals.
The following sections address these aspects to provide a comprehensive overview of NGS and the current status of other omics technologies applied to diagnosis in clinical practice.
Omics technologies and their integration in clinical practiceOmics technologies are a set of methods used to comprehensively study all the components involved in the molecular processes of living organisms.
All of these technologies generate massive volumes of data that need to be analyzed using complex statistics-based methods. They also require high-capacity computing infrastructure, information storage and management resources and specialized personnel for analysis and governance.
In the context of diagnosis, the most widely used omics technologies are genomics, transcriptomics, proteomics, metabolomics and epigenomics.
Of these, genomic technologies, which are used to analyze the DNA of human individuals (such as NGS), are the most widely applied in routine clinical practice. The use of other omics technologies is, for the most part, limited to research with the ultimate goal of translating the findings to clinical practice. We present some examples of these applications later in the text.
Massive sequencing as a diagnostic techniqueNext generation sequencing is considered a diagnostic technique, and, over the past decade, molecular assays based on this technology have been integrated into the service portfolio of the public health care system in Spain.
Currently, the implementation of NGS methods is uneven across autonomous communities and partly determined by the technological resources and staff trained in bioinformatics required to ensure the validity of the findings.
Their introduction in molecular laboratories has marked a paradigm shift: in the past, technicians performed the experiments and clinicians issued the report. Now, NGS requires data analysis prior to clinical interpretation, which in turns requires that laboratories adapt to meet these new demands.
The main distinctive features of NGS as a diagnostic technique can be summarized as (1) decentralization and (2) the lack of built-in quality control checkpoints.
Decentralization involves the execution of multiple sequential steps to obtain a clinically interpretable result that may be performed by different laboratories or institutions. Each step requires specific facilities, specialized resources and qualified staff.
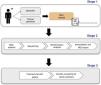
This workflow (Fig. 1) begins with the physician ordering the test (after obtaining written informed consent), continues with DNA extraction from the tissue of interest and the creation of DNA fragment libraries using standardized protocols, proceeds to DNA sequencing on a high‑throughput sequencing platform and culminates with a bioinformatics analysis.

Flow chart of the NGS workflow.
The stages involved in performing an NGS test are outlined below: first, the patient visits the facility and the requesting physician (specialist or clinical geneticist) orders an NGS test after obtaining signed informed consent (stage 1). The test is then carried out sequentially (stage 2) so that, finally, the results in the NGS report can be communicated to the patient in the office and, if indicated, genetic counseling can be offered to other family members (phase 3).
Subsequently, a clinical specialist prepares an NGS report that includes the findings of the test (positive, negative or inconclusive) and other relevant information about it.
Decentralization allows the entire test or some of its phases to be outsourced, giving rise to the following implementation models: (a) in-house, (b) outsourced, or (c) hybrid.
The fragmentation of the workflow into subprocesses requires standardized communication protocols between all parties to ensure traceability and correct management of the diagnostic process. Process oversight and monitoring are essential to ensure analytical and clinical validity,8 even in outsourced or hybrid implementation models.
The overall validity of the test largely depends on the analytical process, as it is during the bioinformatics analysis that the majority of issues (such as sample swaps, contamination or low-quality samples) are identified, which ultimately helps determine which samples are suitable for diagnosis.
However, one of the challenges posed by NGS is the lack of built-in quality control checkpoints beyond those present in the sequencing platforms. This is of critical importance, especially in variant calling, where poorly calibrated pipelines or incomplete raw sequencing output can generate files with deficient information.
Furthermore, the lack of standardized best practices and decentralization (in cases where several laboratories are involved in performing the test) complicate the implementation of corrective measures that ensure quality and process integrity. This underscores the importance of laboratories adopting quality frameworks, such as ISO standards, to ensure patient safety. We will address these issues later in the text.
Another important aspect of NGS is the ability to perform targeted sequencing using disease-specific gene panels, focusing on specific genomic regions to reduce costs and analysis time. Similarly, when broader analysis is required, whole exome sequencing (WES) and whole genome sequencing (WGS) are possible alternatives.
Last of all, we must highlight the need to manage the large data volumes generated by each test. The generated file size varies according to multiple factors, such as the size of the sequenced region, the depth of coverage (also referred to as read depth), and the number of samples sequenced simultaneously.
For instance, the specifications for the Illumina NovaSeq 6000 system9 provide a general idea of the magnitude of the files that are generated in these processes: run output folders range from approximately 230 gigabytes (GB) to 3 terabytes (TB), whereas a complete test, considering all the files generated during both sequencing and analysis, may add up to between 560 GB and 6.5 TB of storage.
NGS data must be treated as medical data, just like medical imaging data or any other clinical information. From this perspective, and in accordance with Law 41/2002 on Patient Autonomy, they must be retained for a minimum of 5 years.
Therefore, having an adequate storage infrastructure is essential, along with efficient planning to enable medium‑term retention of genomic data.
Other omics applied to diagnosisIn addition to genomics, other omics technologies are gaining relevance in clinical practice. Although they are already sometimes used for diagnostic purposes, their application remains largely confined to preclinical research.
These technologies play a key role in the discovery, analysis and validation of new therapies, biomarkers and diagnostic tools, which are fundamental to the advancement of personalized medicine.
In recent years, there have been significant advances in proteomics, which analyzes the presence of proteins in a sample on a large scale, thanks to technological improvements and more effective bioinformatics analysis.10 One salient example is the Overa test, developed by Vermillion, which uses tandem mass spectrometry (LC-MS/MS) to assess the risk of ovarian cancer in women with pelvic masses. This test was approved by the United States Food and Drug Administration (FDA) in 2016.
The analysis of metabolites is already an established practice in pediatric care, especially in the context of newborn screening for congenital metabolic disorders. Metabolomics constitutes a significant advance by enabling the untargeted analysis of thousands of metabolites, facilitating the identification of metabolic signatures in fluids and tissues. Research is currently underway to apply metabolomics to diseases in which early detection could improve patient outcomes, such as type 2 diabetes.11
Another field of interest is precision oncology, where identifying biomarkers is essential to enable early detection, tumor classification and treatment response monitoring. In this regard, epigenomic analysis has provided notable success stories, such as the use of EGFR‑activating mutations as biomarkers, which has significantly improved survival in patients with non‑small-cell lung cancer (NSCLC).12
In the field of molecular genetics, it is worth noting the advent of multiomics integration methodologies, which enable integration of information to improve the diagnosis of patients who exhibit characteristic phenotypes but in whom the causal disease mechanism has not yet been identified.
Thus, the inclusion of RNA sequencing (RNA‑seq) data in molecular studies allows analysis of the patient’s transcriptome, facilitating the identification of aberrant splicing and gene expression.13
Epigenetic signature analysis14 is also proving useful in patients with neurodevelopmental disorders, as it allows classification of patients with complex syndromic presentations or differentiation of overlapping phenotypes, such as Kabuki syndrome and CHARGE syndrome, thereby improving diagnostic precision.
Analysis of NGS data: bioinformatics pipelinesA bioinformatics pipeline is the set of tools and algorithms used to analyze the raw output from sequencing platforms and identify the genetic variants carried by the patient. The English technical term “pipeline” is widely used in diagnostic laboratories in Spain, as are other bioinformatics terms that have been adopted in their untranslated version.
Pipelines typically include free and open‑source software, although there are also proprietary pipelines and pipelines that incorporate commercial tools as part of the workflow.
Pipelines must be tailored to the type of variant and to whether it is of germline or somatic origin, using specific analysis methods in each case.
In germline variant analysis, the main classes of variants usually characterized are (1) single nucleotide variants (SNVs), (2) small insertions or deletions (INDELs), (3) copy number variations (CNVs), and (4) other structural variants (SVs).
The need to tailor pipelines and their configuration to each specific use often leads to in‑house pipelines being chosen over commercial pipelines. In‑house pipelines are developed within laboratories and are more flexible, but their use raises regulatory and quality concerns, whereas commercial pipelines offer other advantages and are validated for specific applications. Table 1 summarizes the pros and cons of both approaches.
Next-generation sequencing implementation models.
| NGS implementation models | |||
|---|---|---|---|
| Model | Description | Pros | Contras |
| In-house | The institution performs the entire NGS workflow in-house, from specimen processing, sequencing, bioinformatics analysis, to data interpretation | • Flexible model that allows adaptation of the workflow to the service portfolio | • Need for qualified/specialized human resources |
| • Control over the process, allowing detection of incidents and implementation of corrective measures | • Need to ensure pipelines adhere to EU regulatory framework | ||
| • Allows innovation and improvements to the workflow | • Costs associated with maintenance of necessary human and material resources | ||
| • Possibility for secondary utilization of the data due to the control over the information | |||
| Outsourced | The institution delegates the entire NGS workflow to a third party. DNA extraction and sample preparation are usually performed in-house | • Reduction in payroll costs | • Lack of flexibility to implement improvements adapted to the needs of the institution |
| • Reduction in equipment acquisition and maintenance costs | • Restrictions to patient data sharing imposed by the GDPR limit the interpretation of the data | ||
| • Reduced investment/effort in accreditation, certification and compliance | • Loss of control over traceability, which poses barriers to sample identification | ||
| • Reports may not fit the institution’s standards, protocols or templates | |||
| Hybrid | The institutions delegates some of the steps of the NGS workflow to a third party. The most common scenario is that the bioinformatics analysis is outsourced to a third party or performed with proprietary software. In this case, the institution performs the interpretation of the data | • Proprietary pipelines need to be accredited/certified to ensure validation and compliance with regulations | • Traceability may be interrupted if the bioinformatics analysis files are not shared by the third party |
| • Reduced investment in innovation at the level of data analysis | • If the institution does not have control over the data, secondary utilization is not possible | ||
| • Reduced need for bioinformatics specialist translated to reduced payroll costs | • If the institution does not have bioinformatics specialists on staff, it is not possible to assess the validity of the results reported by a third party | ||
| • In some cases, the institution is not able to determine the occurrence of incidents in the workflow or to identify samples | |||
| • Rigidity in analytical processes applied to specific needs or cases | |||
Abbreviations: GDPR, General Data Protection Regulation; NGS, next-generation sequencing.
At present, in hospital settings, there are three broad NGS testing implementation models: in-house (in which the institution performs every step of the test), outsourced, also known as send-out or externalized (the service is outsourced to a third party) and hybrid (when only some of the steps of the test are outsourced to a third party). The table details the pros and cons of each of these models.
The following sections describe the logic of pipeline analysis. Tables 2 to 5 summarize the file formats generated in each step in addition to the tools most widely used for each purpose, including the corresponding sources.
File formats in the bioinformatics analysis workflow.
| File type | Information contained | Source URL |
|---|---|---|
| FASTQ | Unaligned DNA sequencing reads | https://en.wikipedia.org/wiki/FASTQ_format |
| SAM | DNA sequencing reads aligned/mapped to a reference genome in text format | https://samtools.github.io/hts-specs/SAMv1.pdf |
| BAM | DNA sequencing reads aligned/mapped to a reference genome in binary format | https://samtools.github.io/hts-specs/SAMv1.pdf |
| CRAM | Compressed file format for DNA sequencing reads mapped to a reference genome | https://samtools.github.io/hts-specs/CRAMv3.pdf |
| GVCF | Intermediate genomic variant call file containing the positions of the detected potential variants | https://sites.google.com/a/broadinstitute.org/legacy-gatk-documentation/frequently-asked-questions/4017-What-is-a-GVCF-and-how-is-it-different-from-a-regular-VCF |
| VCF | File containing the detected variants | https://samtools.github.io/hts-specs/VCFv4.2.pdf |
Abbreviations: VCF, List of the different file formats generated in a bioinformatics analysis. For each format, the table describes the contents of the files and includes a URL to access an in-depth description of its specifications.
Tools used in the initial stage of a bioinformatics pipeline.
| Name | Step | URL |
|---|---|---|
| bcl2fastq (Illumina) | Demultiplexing | https://emea.support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html |
| Trimmomatic | Trimming | http://www.usadellab.org/cms/?page=trimmomatic |
| Cutadapt | Trimming | https://cutadapt.readthedocs.io/en/stable/ |
| BBDuk | Trimming | https://github.com/BioInfoTools/BBMap |
| BWA-MEM | Alignment | https://github.com/lh3/bwa |
| Bowtie2 | Alignment | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Minimap2 | Alignment | https://github.com/lh3/minimap2 |
| Picard | General suite | https://broadinstitute.github.io/picard/ |
| Samtools | General suite | http://www.htslib.org/ |
The initial stage includes the following core steps: (a) trimming, (b) alignment and (c) filtering of duplicates. The table lists the most widely used tools to perform these tasks along with the URL for the software.
Tools used for detection of variants.
| Name | Type of variant | Type of test | URL |
|---|---|---|---|
| GATK4/ | SNVs/INDELs | panel/WES/WGS | https://gatk.broadinstitute.org/hc/en-us |
| DeepVariant | SNVs/INDELs | panel/WES/WGS | https://github.com/google/deepvariant |
| DECON | CNVs | panel/WES | https://github.com/RahmanTeam/DECoN |
| XHMM | CNVs | panel/WES | https://github.com/RRafiee/XHMM |
| panelcn.MOPS | CNVs | panel/WES | https://github.com/bioinf-jku/panelcn.mops |
| ExomeDepth | CNVs | panel/WES | https://github.com/vplagnol/ExomeDepth |
| CODEX2 | CNVs | panel/WES | https://github.com/yuchaojiang/CODEX2 |
| Genome STRiP | SVs | WGS | https://software.broadinstitute.org/software/genomestrip |
| Delly | SVs | WGS | https://github.com/dellytools/delly |
| Manta | SVs | WGS | https://github.com/Illumina/manta |
| LUMPY | SVs | WGS | https://github.com/arq5x/lumpy-sv |
| ExpansionHunter | Repeat expansions | WES/WGS | https://github.com/Illumina/ExpansionHunter |
| GangSTR | Repeat expansions | WES/WGS | https://github.com/gymreklab/GangSTR |
Abbreviations: CNV, copy number variation; INDELs, small insertions and deletions; SNV, single nucleotide variant; SV, structural variant; WES, whole exome sequencing; WGS, whole genome sequencing.
List of the most widely used tools for identification of variants in aligned reads. The variants that can be detected with these tools are SNVs, INDELs, CNVs and other SVs.
Tools use for variant annotation.
| Name | Type of variant | URL | |
|---|---|---|---|
| Tools for annotation and interpretation | |||
| Annovar | SNVs/INDELs | https://annovar.openbioinformatics.org/en/latest/ | |
| snpEff | SNVs/INDELs | http://pcingola.github.io/SnpEff/ | |
| VEP | SNVs/INDELs | https://github.com/Ensembl/ensembl-vep | |
| ClassifyCNV | CNVs | https://github.com/Genotek/ClassifyCNV | |
| CharGer | SVs | https://github.com/ding-lab/CharGer | |
| AnnotSV | SVs | https://lbgi.fr/AnnotSV/ | |
| Population databases | |||
| GnomAD v4 | SNVs/INDEL/SVs | https://gnomad.broadinstitute.org/news/2023-11-gnomad-v4-0/ | |
| others | SNVs/INDELs | https://www.ensembl.org/info/genome/variation/species/populations.html | |
| Pathogenicity predictors | |||
| CADD | SNVs/INDELs | synonymous, missense, nonsense, frameshift, splicing, noncoding, promotor and enhancer variants | https://cadd.bihealth.org/ |
| FATHMM | SNVs/INDELs | synonymous, missense, nonsense y frameshift | http://fathmm.biocompute.org.uk/ |
| AlphaMissense | SNVs | missense | https://alphamissense.hegelab.org/ |
| SpliceAI | SNV/INDELs | splicing | https://github.com/Illumina/SpliceAI |
| X-CNV | CNVs | – | http://119.3.41.228/XCNV/index.php |
| CADD-SV | SVs | – | https://cadd-sv.bihealth.org/ |
| Curated databases of known variants accessible to pipelines | |||
| ClinVar | SNVs/INDEL/SVs | https://www.ncbi.nlm.nih.gov/clinvar/ | |
| HGMD | SNVs/INDEL/SVs | https://digitalinsights.qiagen.com/hgmd-spanish/ | |
| dbSNP | SNVs/INDELs, insertions, retrotransposons and microsatellites | https://www.ncbi.nlm.nih.gov/snp/ | |
| ClinSV | SVs | https://github.com/KCCG/ClinSV | |
Abbreviations: CNV, copy number variation; INDELs, small insertions and deletions; SNV, single nucleotide variant; SV, structural variant.
The table shows the best-known tools and resources used for performing this step. It starts with some of the most widely used annotation programs, listed with their URLs for accessing the software. Then, it lists the most used databases used for establishing the allele frequency of variants. Next, it lists a series of pathogenicity predictors with their corresponding URLs. Last of all, it includes some databases of known variants whose effects have been reviewed by experts and which can be accessed by bioinformatics pipelines.
Fig. 2B presents the typical workflow of a pipeline. There is usually an initial stage that includes (1) processing the sequencing output, (2) filtering the data, and (3) aligning DNA reads to a reference genome.
On Illumina sequencing platforms, the primary output from the sequencer must undergo a demultiplexing step. The purpose of this process is to separate the raw data generated by the instrument into individual samples and assign each sample its corresponding FASTQ files, which contain the DNA sequence reads (hereafter, “reads”). When sequencing is performed in paired‑end mode, reads are obtained from both ends of each DNA fragment, which results in two FASTQ files per sample.
Next, a trimming step is applied to remove low‑quality reads, sequencing artifacts, and adapter sequences. Each trimmed read is then aligned to a reference genome, and the choice of reference genome can affect variant calling,15 so it is an important aspect to consider.
Aligned reads are stored in BAM (binary alignment/map) files, which can be opened and viewed in genome browsers. Manual review of regions of interest in these viewers provides experts with additional, context‑rich information that complements automated analyses.
Each read has an associated quality score that depends on multiple factors. For example, in short‑read sequencing, low‑complexity or repetitive genomic regions are typically covered by lower‑quality alignments, which can lead to coverage bias and hinder variant detection.
Identification of variantsAfter mapping each DNA read to the reference genome, genetic variants are identified in a process known as variant calling. This step is highly specialized, because the algorithms used differ based on the type of variant that is to be detected.
These algorithms are based on statistical methods and require an adequate read depth to identify patterns. Fig. 2A shows a single nucleotide variant (SNV) in the AGL gene.

Summary of the bioinformatics analysis process.
(A) Example of a heterozygous SNV in the AGL gene. Aligned reads are shown in grey and the nucleotide sequence of the featured exon fragment can be seen at the bottom. (B) Sequential steps performed in the bioinformatics analysis process that must be implemented through a pipeline. (C) Information for the variant shown in (A) with the corresponding annotations provided to support the interpretation of the variant.
In addition to identifying the variant, it is essential to determine whether it is heterozygous or homozygous (that is, whether the variant is present in one or both alleles). However, the sequencing workflow generally does not preserve information about the allelic origin of each read, so this must be inferred later in the process.
Variant calls are subject to uncertainty of varying degree, which may give rise to false positives. Therefore, the reliability of the findings must be evaluated using the quality scores provided by the available tools, taking into account biases that may affect specific genomic regions. In addition, viewing variants in their genomic context is recommended for a more accurate interpretation.
SNV calling algorithms have evolved over the past decade, and samples are now analyzed in groups rather than individually to minimize the impact of recurrent artifacts within sequencing batches (joint calling).
GATK416 is the best-known toolkit for the purpose, while DeepVariant,17 which is based on neural networks, can be applied in different contexts, including somatic variant calling. An article published by Barbitoff et al. (2022)18 offers an overview of bioinformatics tools for calling SNVs and INDELs.
To detect CNVs, the most common method estimates gene dosage from normalized read depth relative to a control population, an approach frequently used in targeted sequencing assays. In whole‑genome sequencing (WGS), multiple strategies are combined to identify CNVs and structural variants such as large deletions, duplications, inversions and translocations.19
Other types of variants that are of interest, especially in hereditary neurodegenerative diseases, are tandem repeats and trinucleotide repeats. To learn more about their detection by NGS, we recommend the review published by the Genomics England Research Consortium.20
Unlike SNVs/INDELs, the detection of SVs using short-read platforms is mainly limited to screening due to the high sensitivity and low specificity of the algorithms, which result in a high false-positive rate. This limitation arises from technological constraints and designs limited to exon sequencing to maximize cost-effectiveness. In clinical practice, hybrid approaches combining multiple technologies are currently being explored following promising results in research.
It is recommended that CNVs and SVs detected by short-read sequencing be confirmed through an orthogonal method. However, this is not always feasible due to the lack of commercial MLPA kits (MRC Holland) or CGH/SNPs arrays covering the regions of interest. Therefore, it is crucial that the reportable range is defined in the test development phase, excluding regions for which there is no clear validation method.
AnnotationIn this step, additional functional, population and clinical information (a process known as annotation) is provided for the identified variants to support their interpretation.
The standard HGVS nomenclature21 should be used to describe SNVs and INDELs based on a carefully selected transcript.22,23 Variant annotation can change based on the selected transcript or transcripts and could affect the accuracy of diagnosis.
Functional data has to be supported with in silico predictions using software applications designed to estimate the probability that an SNV or INDEL has a deleterious impact.24Table 5 lists the most widely used predictors.
Population-scale aggregated data helps identify variants in the reference population. The gnomAD database25 is one of the most comprehensive resources, as it has data for 195 000 individuals that serve as a reference population.
Annotation should also include information on known expert-curated, clinically important variants, available through databases such as dbSNP, ClinVar and HGMD (the latter requires a license for use).
Although the workflow for SNVs and INDELs has been standardized, tools for CNV and SV annotation remain limited and lack widely accepted standards. The use of tools such as ClassifyCNV and AnnotSV, as well as CharGer, which incorporates criteria from the American College of Medical Genetics and Genomics and Association of Molecular Pathology (ACMG/AMP),26 considered the standard framework for variant classification, should be considered.
Filtering, prioritization and interpretationSince there is no clear definition of the role and scope of practice of clinical bioinformaticians, there are differing views on who should be responsible for variant filtering and prioritization.
This step should preferably be performed by experts in molecular genetics, who would be responsible for determining the pathogenicity of candidate variants according to the ACMG/AMP guidelines.
However, more complex analyses require the use of virtual gene panels and/or advanced filtering with specialized or custom-made algorithms. Therefore, close collaboration between bioinformaticians and molecular geneticists is essential to support decision-making in the final phase of clinical NGS.
Various filtering and prioritization tools are available to facilitate these tasks; most require a license, allowing handling of extensive lists of variants, although their description is beyond the scope of this review.
Other considerationsAnalysis methods are constantly evolving, so it is essential to upgrade pipelines with more effective algorithms and updated databases. Since there are no regulations governing the frequency of these updates, it is crucial to document pipeline versions including detailed descriptions of the software and resources used, and to include this information in NGS reports to trace the analysis process.
Pipelines need to be monitored to assess their performance over time and ensure their validity when changes are introduced in the diagnostic process. Also, in the case of in-house pipelines, it is essential that program parameters are configured correctly in the development phase.
Therefore, well-characterized reference samples, such as those provided by the Genome in a Bottle (GIAB) consortium,27 need to be used to optimize performance and assess the reproducibility of the results. However, reference data are not available for some alterations, which limits the implementation of well‑calibrated algorithms in routine diagnostic practice.
For instance, in the case of the GIAB NA12878/HG001 reference genome, vials with DNA samples of this particular individual can be obtained to validate the bioinformatics process alone or the entire workflow from the initial experimental stages.
Regular evaluations can also be performed to assess performance against a standard or independent review, for instance, using EMQN external quality assessment schemes. Implementation of evaluations by external agencies is recommended to assess the performance of pipelines.
The reproducibility of the results obtained with the pipelines may be affected by the hardware and the software versions of the computational tools used in the analysis. For this reason, running pipelines in containers is recommended,28 as containers package the code along with all necessary dependencies and archives, guaranteeing consistency across environments and improving portability, traceability and reproducibility.
Quality assuranceThe analysis pipeline must be complemented by quality assurance processes. The primary objective is to determine which samples are adequate for diagnosis and, in the case of inadequate samples, to identify the reason for it (whether it is due to an incident in the process or other factors). Quality assessment tools also provide information on the effectiveness of the testing process.
It must be taken into account that quality metrics are not standardized and, to date, there is no consensus on the metrics that should be analyzed and included in reports on a mandatory basis. A detailed description of the most commonly used metrics is beyond the scope of this article, as it would require a more detailed and comprehensive discussion. Nevertheless, Table 6 presents a selection of quality aspects that should be considered both for process monitoring and sample review.
Aspects to consider for quality control purposes in the evaluation of NGS tests and assessment of sample adequacy.
| Quality aspect | Bioinformatics analysis | Molecular analysis |
|---|---|---|
| Establishing the sensitivity and specificity of the analysis | Yes, with reference samples. | Yes, using external and accredited validation schemes (EMQN). |
| This information should be included in the NGS report | This information should be included in the NGS report | |
| Establishing the reproducibility of performance metrics | Yes, with periodic monitoring and comparisons with external laboratories | Yes, with double-blind analysis of the same sample and comparison with external laboratories |
| Establishing the reliability of performance metrics | Yes, using different reference samples | Yes, at regular intervals, with double-blind analysis and comparison with external laboratories using different samples and panels |
| Establishing the reproducibility and traceability of the analysis process for a given sample | Yes, with containerized pipelines and a data structure allowing storage of data for the entire testing process. | Yes, with an agreed upon and documented prioritization protocol. Back-up copy protocol for the protocolized steps to enable traceability of decision-making |
| This information must be included in the NGS report | ||
| Establishing the clinical validity and diagnostic utility of the test | – | Yes, using external and accredited validation schemes (EMQN). |
| Apply quality control measures to each of the samples. | Yes. These measures must be documented. The limits of normal must be established based on robust statistical analyses. | Molecular geneticists must be familiar with the metrics or control measures applied to each sample and must be qualified to understand them. |
| Each sample should be labeled as “adequate”, “inadequate” or “adequate but subject to incidents” for the purpose of diagnosis | When asked about the results for an “inadequate” sample, they must be able provide objective reasons for the delay to the requesting physician. | |
| Variants must be confirmed or, when that is not possible, methods to minimize false positives must be implemented. | ||
| Standardize the criteria and format for molecular/genetic test reports | Yes. The Bioinformatics Unit must participate in defining and the relevant information to be included in the report and the format or template for the report. | Yes. Molecular geneticists must establish general criteria, analysis protocols and tools to facilitate the work. |
| Standardized criteria must be established for the reporting incidental findings. | ||
| The NGS report must be standardized and must include all possible tests or uses of these techniques. | ||
| Implement standard quality assurance frameworks in accordance with a quality standard or ISO and achieve certification/accreditation. | Yes. The Bioinformatics Unit must participate in quality assurance. If the implementation model is hybrid or outsourced, the quality processes must also be adapted to comply with ISO standards | Yes. Molecular geneticists must participate in quality assurance. If the implementation model is hybrid or outsourced, the quality processes must also be adapted to comply with ISO standards |
| UNE-EN accreditation of laboratory | Yes. The Bioinformatics Unit must comply with standards certifying the validity of the analysis process, whether it is outsourced or not. | Yes. Molecular geneticists must comply with standards certifying the validity of the analysis process, whether outsourced or not |
List of aspects to consider in a quality assurance process across the bioinformatics analysis stage, variant interpretation in molecular analysis stage and the writing of the report for the patient.
There are limitations to short-read sequencing due to the small size of the fragments, which results in ambiguous alignments in certain regions. This particularly affects fragments with low complexity (homopolymers), repetitive regions (tandem repeats), or high homology (segmental duplications and pseudogenes). As a result, it is difficult to determine the location of the reads accurately, especially when repetitive regions exceed the fragment size.
Alignment algorithms map each read to multiple locations, reducing quality and skewing variant identification, producing artifactual variants or failing to identify existing variants. One example is the PKD1 gene, crucial for the diagnosis of polycystic kidney disease, where the presence of pseudogenes drastically reduces the number of variants identified by short-read sequencing, calling for the use of more advanced methods.29
New approaches, such as linked-read or long-read sequencing, are proving useful in the interrogation of certain regions that have been challenging with other sequencing methods, for instance, the region encoding the human leukocyte antigen (HLA) complex.30
To overcome the limitations of established diagnostic methods, it is necessary to explore new approaches and integrate new omics technologies, as discussed in previous sections.
NGS data useGenomic data should be treated as medical data and, therefore, should be managed by the same parties responsible for health care data governance and stewardship in health care systems.
Reanalysis due to changes in the suspected diagnosis, new cases in the family or the reuse of variant data requires that the data continue to be available. For this reason, archiving systems must guarantee access to the data during the legally mandated period and beyond.
The creation of patient registries facilitates the interpretation of candidate variants, as their presence/absence in the series allows their classifications to be established with greater confidence. Queries across datasets can be performed using the Beacon31 data discovery protocol, which also allows the incorporation of clinical data on the patient.
Secondary use of NGS data is essential to feed the research-development-implementation loop and allow laboratories to incorporate technological advances into their practices, as long as the informed consent allows reuse of the data.
Finally, it is important to anonymize NGS records to comply with the General Data Protection Regulation (GDPR), avoiding the inclusion of patients’ personal information.
Regulatory aspectsSince 2014, Eurogentest has issued recommendations to improve the accuracy of genetic testing, such as the guidelines for diagnostic NGS.32 In Europe, NEQAS (United Kingdom) and VKGL (Netherlands) are benchmarks for good practice in NGS, given the absence of a regulatory framework. However, ISO 15189 accreditation is widespread in NGS laboratories within Europe.
In-house pipelines are considered in vitro diagnostic medical devices and fall under the scope of Regulation (EU) 2017/746. In Spain, at the domestic level, new, updated legislation is being drafted to replace Royal Decree 1662/2000, and a European working group has published a guidance document on the subject.33
If third-party services are used to store data in the cloud, the third party must provide proof of compliance with its responsibilities in personal and health care data handing and management according to the GDPR.34
DiscussionThe bioinformatic analysis of NGS data is complex and requires specialized personnel, adequate infrastructure and training of medical staff to correctly interpret the results.
Oversight and quality control are essential to ensure the analytical and clinical validity of the test. This is particularly relevant in cases where bioinformatic analysis is outsourced to a third party.
Although short-read technologies are very efficient and achieve a high diagnostic yield, emerging technologies need to be integrated to address those cases in which a diagnosis cannot be reached due to, among other possible causes, an unknown genetic basis or a complex clinical presentation, pushing beyond the limitations of current diagnostic methods.
Regulatory frameworks are key, but progress still needs to be made toward the development and implementation of standardized practices established by expert consensus. In this regard, administrations must be more proactive in promoting standardization and best practices in the application of omics to clinical practice.
Declaration of Generative AI and AI-assisted technologies in the writing processDuring the preparation of this work, the author used ChatGPT to improve the readability of the manuscript she had written. After using this tool/service, the author reviewed and edited the content as needed and takes full responsibility for the content of the publication.
The authors have no conflicts of interest to declare.
