



Inborn errors of metabolism are a highly heterogeneous group of orphan diseases. Diet therapy and enzyme and coenzyme replacement are the most frequently used treatment. There are few patients and published studies about inborn errors of metabolism. The main objective of this study was to describe the effectiveness of orphan drugs in inborn errors of metabolism in paediatric patients.
Material and methodsRetrospective descriptive study of 24 months on patients diagnosed with inborn errors of metabolism during childhood and who attended the pharmacy clinic or Day-Care Unit of a 630-bed general hospital.
ResultsThe study included 15 patients with a median age of 17.8 years and were treated with nine different drugs: sapropterin, sodium phenylbutyrate, miglustat, velaglucerase, sebelipase, idursulfase, 5-hydroxytryptophan, succinate, and riboflavin. Seven different inborn errors of metabolism were observed: phenylketonuria, defects of the urea cycle, Gaucher, Nieman-Pick, Hunter’s disease, along with acid lipase deficiency, and mitochondrial diseases. Orphan drugs used for the treatment of inborn errors of metabolism accounted for 1.3% of hospital drug costs. Some orphan drugs achieved asymptomatic patients, but others just produced a modest symptomatic improvement. Most patients showed good tolerance to the treatment.
ConclusionsOrphan drugs used in inborn errors of metabolism had an easy to manage toxicity profile, with many disparities in effectiveness. These drugs have a high economic impact. The cost-effectiveness ratio for orphan drugs is a controversial issue due to their high cost and the inconclusive clinical evidence.
Los errores congénitos del metabolismo son un grupo muy heterogéneo de enfermedades raras. La mayoría se pueden tratar con dieta y sustitución enzimática.
Existen pocos pacientes y pocos estudios publicados en estas enfermedades. Por ello, se ha llevado a cabo un estudio con el objetivo de evaluar la efectividad de los medicamentos huérfanos utilizados en errores congénitos del metabolismo de un hospital general de 630 camas.
Material y métodosEstudio descriptivo restrospectivo de 24 meses de duración en un hospital general de 630 camas. Se incluyeron los pacientes diagnosticados durante la infancia de errores congénitos del metabolismo y que acudieron a Hospital de Día o a la consulta de Farmacia.
ResultadosSe estudiaron 15 pacientes, con una edad media de 17,8 años, que recibieron tratamiento con 9 fármacos: sapropterina, fenilbutirato de sodio, miglustat, velaglucerasa, sebelipasa, idursulfasa, hidroxitriptófano, succinato y riboflavina. Presentaban 7 errores congénitos del metabolismo diferentes: fenilcetonuria, trastorno del ciclo de la urea, Gaucher, Niemann-Pick, Hunter, déficit de lipasa ácida liposomal y defectos mitocondriales. Los medicamentos huérfanos para estos trastornos supusieron el 1,3% del gasto de farmacia. Algunos medicamentos huérfanos lograron que los pacientes estuvieran asintomáticos y otros, sólo produjeron una discreta mejoría. La efectividad de las fórmulas magistrales se evaluó mediante criterios subjetivos. En general, los pacientes presentaron buena tolerancia a estos tratamientos.
ConclusionesLos medicamentos huérfanos utilizados para errores congénitos del metabolismo presentan buen perfil de seguridad pero gran disparidad en cuanto a su efectividad. Suponen un alto impacto económico.
La incertidumbre en cuanto a la evidencia clínica de los medicamentos huérfanos junto con el elevado coste, hace que la relación coste-efectividad sea controvertida.
Rare diseases (RDs) are diseases with a prevalence of less than 5 cases per 10 000 inhabitants in the European Union. Most cases have onset in childhood due to the high frequency of genetic disorders and congenital anomalies. However, the prevalence is higher in adults than in children due to the high mortality of some severe childhood diseases and the contribution of diseases with later onset.1
Congenital errors of metabolism (CEMs) are a broad and heterogeneous group of RDs, including more than 500 types. They usually have onset early in life, and their overall prevalence is of 1 case in 600–1000 births.
Most CEMs are autosomal recessive genetic disorders and due to a change in the structure of function of a protein.2
Generally, CEMs are treated with dietary measures, orphan drugs for enzyme replacement therapy, or both. The European Commission defines orphan drugs as medicinal products for treatment of life-threatening or chronically debilitating conditions with a prevalence of no more than 5 cases per 10 000 inhabitants or for which there is no satisfactory method of diagnosis, prevention or treatment authorised in the European Union.3,4
The low prevalence of these diseases, the geographical dispersion of cases and the scarcity of the literature on the subject, in addition to the methodological limitations of some of the published studies, hinder our understanding of rare diseases.
In this context, researching rare diseases in clinical practice seems important. The primary objective of our study was to assess the effectiveness of the orphan drugs used for treatment of CEMs diagnosed in paediatric patients. The secondary objectives were to analyse the safety profile and economic impact of these drugs.
Material and methodsWe conducted a retrospective, observational and descriptive study in a general hospital with 630 beds between 2018 and 2019. We included every patient with a CEM diagnosed during childhood managed with orphan drugs through the outpatient pharmacy department or in the day hospital.
We collected data on demographic characteristics (age, sex and CEM) and clinical variables related to disease outcomes.
The primary outcome of the study was the effectiveness of orphan drugs. We evaluated the effectiveness based on data from the clinical trials of each drug:
- •
Sapropterin: reduction in plasma phenylalanine (<360 nmol/mL) and improved tolerance of phenylalanine intake.5
- •
Sodium phenylbutyrate: reduction of plasma ammonium (19−82 µg/dL) and decreased frequency of episodes of hyperammonaemia.6
- •
Velaglucerase: normalization of cell counts (platelets and haemoglobin), visceral organ volumes and marker levels, and reduction of bone infiltration.7
- •
Miglustat for Gaucher disease: reduction in hepatosplenomegaly.8
- •
Miglustat for Niemann-Pick disease type C: slowing the progression of disease, stabilization or improvement of dysphagia and ambulation impairment.8
- •
Idursulfase: reduction in glycosaminoglycans (GAGs) in urine and visceral organ enlargement, improved endurance (6-min walk test [6MWT]) and lung function (forced vital capacity [FVC]).9
- •
Sebelipase alfa: improvement in lipid profile and liver enzyme levels.10
- •
Compounded preparations for mitochondrial respiratory chain defects: since these drugs have not undergone the marketing authorization process or clinical trials, we applied clinical criteria to assess their effectiveness.
As secondary outcomes, we collected information on the safety of orphan drugs and the cost of annual treatment.
We retrieved data from the pharmacy dispensation and electronic health record databases.
We adhered to the ethical principles for medical research in human subjects established in the Declaration of Helsinki.11 Also, in adherence with Organic Law 3/2018 of December 5 on the protection of personal data and safeguarding of digital rights, we protected the confidentiality of the patients in data collection and handling.12 The study was approved by the Ethics Committee on Research with Medicinal Products of La Rioja.
ResultsWe selected 15 patients, 60% male, with a median age of 17.8 years (3–49) with 7 different CEMs. The orphan drug used most frequently was sapropterin (n = 5; 33.0%), prescribed for the treatment of phenylketonuria. Table 1 presents the main characteristics of the CEMs.13–21
Main characteristics of the congenital errors of metabolism found in the study.
| Disease | Enzyme deficiency | Accumulated product | Inheritance | Prevalence | Clinical features | Treatment |
|---|---|---|---|---|---|---|
| Phenylalaninaemia | Phenylalanine hydroxylase ± cofactor BH4 | Phenylalanine | Autosomal recessive | 1/10 000 live births | Intellectual disability, microcephaly, hypsarrhythmia, motor abnormalities, cutaneous lesions | 1. Dietary measures with monitoring of phenylalanine |
| Phenylketonuria | 2. Enzyme replacement therapy (ERT) with tetrahydrobiopterin (BH4) in respondents. | |||||
| In the case of BH4 deficiency, administration of levodopa and 5-hydroxytriptophan precursors | ||||||
| Urea cycle disorder | Enzymes involved in the synthesis of urea | Ammonia | Autosomal recessive Ornithine transcarbamylase deficiency: X-linked | 1/25−50 000 live births | Neonatal forms are nearly always lethal, while late-onset forms have a variable presentation and course. It frequently manifests with abdominal pain and hepatomegaly, growth faltering, psychomotor delay, encephalopathy with behavioural disorders and hallucinations. | In patients with argininosuccinate synthetase and argininosuccinate lyase, it is possible to stimulate the rest of the cycle with administration of arginine. In all other cases, sodium phenylbutyrate is a treatment option. |
| Nieman-Pick type C | Lipid transport protein | Cholesterol and sphingolipids | Autosomal recessive, changes in NPC1 and NPC2 | 1−9/100 000 births | Hepatosplenomegaly and severe central nervous system involvement with psychiatric symptoms | Miglustat was approved in 2009 for treatment of NPD type C. |
| Gaucher disease | Acid beta-glucosidase | Glucosylceramide | Autosomal recessive | 1/50 000−500 000 | - GD type 1: cytopenia and hepatomegaly. Cardiovascular risk factor. | 1. Enzyme replacement therapy (ERT): imiglucerase and velaglucerase (the latter is only indicated in GD type 1). |
| - GD type 2: acute neurologic form. ERT does not prevent progression, death within 1 year | 2. Substrate reduction therapy (SRT): miglustat and eliglustat. | |||||
| - GD type 3: slowly progressing neurologic symptoms | 3. Stem cell transplantation: can achieve a cure, high risk. | |||||
| Hunter syndrome (MPS type II) | Iduronato-2-sulfatasa | Dermatan sulphate and heparan sulphate (GAGs) | X-linked | 1/72 000−77 000 male births | Hernia, hepatosplenomegaly, short stature, behavioural disorders and psychomotor regression resulting in intellectual disability, hearing loss, cardiac and respiratory disorders. In the attenuated form there is no cognitive impairment | 1. Haematopoietic stem cell transplantation (HSCT) |
| 2. ERT: purified form of the enzyme iduronate-2-sulfatase: Idursulfase | ||||||
| 3. In trials: intrathecal ERT | ||||||
| Lysosomal acid lipase deficiency (LALD) | Lysosomal acid lipase | Cholesteryl esters and triglycerides | Autosomal recessive, changes in LIPA gene | 1/350 000 births | Hepatomegaly and elevation of serum transaminases, LDL and TG, usually low HDL levels. Most develop chronic liver disease, may develop cardiovascular disease | 1. Adjuvant therapy: lipid-lowering drugs or liver transplant. |
| 2. ERT: human recombinant LAL approved in 2015: sebelipase alfa. | ||||||
| Mitochondrial respiratory chain defect | Oxidative phosphorylation system (OXPHOS) | Mitochondrial, autosomal recessive or autosomal dominant. Sporadic in some cases | 1:50 000 | Usually multisystemic disorders. Chiefly involve skeletal muscle, nervous tissue or bone marrow (high O2 consumption). The most frequent manifestations are neuromuscular. | 1. Improve respiratory chain function (riboflavin, ubiquinone) | |
| 2. Facilitate elimination (carnitine, tocopherol). | ||||||
| 3. Prevent oxidative stress (retinol, lipoic acid, vitamin C…). | ||||||
| b) Organ transplant | ||||||
| c) Gene therapy: research | ||||||
| d) Supportive care: dietary intervention, vitamin supplementation |
Five patients with a mean age of 16 years (11–24) received sapropterin. The median dose was 700 mg/day (300–900). The median plasma phenylalanine level was 369.6 ± 157 nmol/mL. Two patients had sustained levels under 360 nmol/mL and were able to have a normal diet without restrictions. The rest required enteral feeding with elimination of phenylalanine and maintained the plasma levels between 400 and 550 nmol/mL.
The patients led a normal life without restrictions, although 2 of them experienced tremors in the extremities.
Urea cycle disorder: glycerol phenylbutyrateA woman aged 34 years had received a diagnosis of ornithine transcarbamylase deficiency at age 11 months. In 2009, she started treatment with sodium phenylbutyrate, switching to glycerol phenylbutyrate at a dose of 3.5 g every 8 h on account of its organoleptic properties in 2019. At the same time, she kept a low-protein diet with citrulline, carnitine, folic acid and calcium supplementation.
Initially, the blood levels of ammonia ranged between 40 and 100 µg/dL, similar to the baseline levels, peaking in 2017. The clinical presentation consisted of severe encephalopathy, self-harm behaviours and autistic features with absence of verbal communication. In the last year the patient had experienced two episodes of epileptic seizures per week, although she exhibited mild improvement in attention and concentration. The most significant adverse effects were flatulence and insomnia.
Gaucher disease (GD): velaglucerase and miglustatTwo male patients, aged 19 and 48 years, had received a diagnosis of GD in childhood. Both led normal, unrestricted lives.
One of them received a diagnosis of GD type 3 shortly after his first birthday, which manifested with hepatosplenomegaly, malnutrition and delays in linear growth, weight gain, walking and communication. The patient received imiglucerase for enzyme replacement therapy (ERT) for 10 years, which due to shortages was switched to velaglucerase at a dose of 60 U/kg every 15 days on compassionate use grounds. The patient also followed a protein-restricted diet. Enzyme replacement succeeded in maintaining haemoglobin and the platelet count in the normal range. The patient reported general malaise following administration of velaglucerase, prompting suspicion of development of antibodies against the enzyme.
The other patient, who had a diagnosis of GD type 1, underwent splenectomy in 1998 and received imiglucerase between 2000 and 2003. In 2007 he enrolled in a clinical trial in which he received miglustat at a dose of 100 mg every 8 h, which he continues to receive at present as substrate reduction therapy. The therapeutic goals were achieved at 1 year of treatment, with a decrease in liver volume greater than 13% and increases in haemoglobin of 0.7 g/dL and in the platelet count 8 × 109/L. The patient experienced hand tremors as an adverse effect of treatment.
Niemann-Pick disease type C: miglustatA girl aged 3 years had a diagnosis of neonatal Niemann-Pick disease type C confirmed by genetic testing. She presented with an enlarged abdominal circumference and bilateral genu valgum. The patient was found to have portal hypertension secondary to cavernous haemangioma, which was treated with spironolactone and propranolol. She started treatment with miglustat after the diagnosis with doses increased gradually to 100 mg/day. The patient developed independent walking at 27 months and language at 3 years, and exhibits adequate social skills. The most recent ultrasound detected splenomegaly that was not clinically significant. The patient experienced diarrhoea and flatulence as side effects of treatment.
Hunter syndrome (HS): idursulfaseTwo boys aged 8 and 9 years received a diagnosis of HS on account of elevated GAG levels in urine and an undetectable/minimal iduronate-2-sulfatase activity. The diagnosis was confirmed by genetic testing, with detection of one de novo mutation and one previously found in the family. Both patients received idursulfase, one at a weekly dose of 12 mg and another in cycling doses of 12 mg, 12 mg and 18 mg delivered weekly. Although there was a marked initial decrease in urine GAG levels when treatment started, the levels subsequently stabilised above the upper limit of normal (Figs. 1 and 2). One of the patients had favourable outcomes in the control of visceromegaly and respiratory function, although these endpoints were assessed using subjective clinical criteria without performance of the 6MWT or spirometry. However, the other patient managed to walk 300 m without shortness of breath in the 6MWT. When it came to the clinical presentation, both patients had learning difficulties with attention deficit, limitations in movement and loss of bladder and bowel control and language skills. One of them had bilateral hearing loss, ametropia and compression neuropathy, while the other had aggressive behaviours, stereotypic movements, sleep disturbances, increased appetite, subclinical mitral and aortic valve insufficiency and kyphosis. He attended a special needs school and engaged in leisure activities without need of respiratory support or feeding or walking aids. In both patients, head imaging tests evinced leukoencephalopathy and supratentorial cortical and subcortical atrophy. Both patients were also receiving intrathecal replacement therapy concomitantly as part of a clinical trial. The adverse effects of treatment were diarrhoea and acral coldness in one, and paroxysmal supraventricular tachycardia in the other.


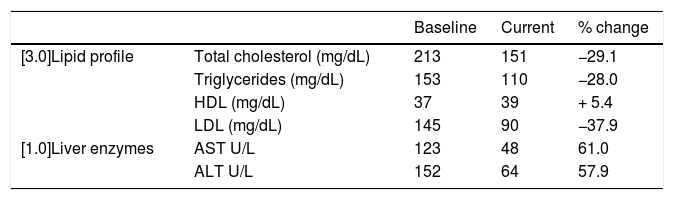
A patient aged 14 years received a genetic diagnosis of LALD in 2016, with identification of double heterozygous mutations. From age 2 years, the patient had faltering growth in weight (8th percentile) and height (30th percentile). He was asymptomatic and had a normal life, with manifestations limited to sinus bradycardia and hepatosplenomegaly with microvesicular steatosis. In 2018 the patient started treatment with sebelipase alfa given every 2 weeks, currently at a dose of 35 mg, which achieved an improvement in the lipid profile and liver enzymes, especially in the past few months (Table 2).
Changes in lipid profile and liver enzymes in a patient with lysosomal acid lipase deficiency.
| Baseline | Current | % change | ||
|---|---|---|---|---|
| [3.0]Lipid profile | Total cholesterol (mg/dL) | 213 | 151 | −29.1 |
| Triglycerides (mg/dL) | 153 | 110 | −28.0 | |
| HDL (mg/dL) | 37 | 39 | + 5.4 | |
| LDL (mg/dL) | 145 | 90 | −37.9 | |
| [1.0]Liver enzymes | AST U/L | 123 | 48 | 61.0 |
| ALT U/L | 152 | 64 | 57.9 |
Two female patients, aged 14 and 23 years, had mitochondrial respiratory chain defects. One had respiratory chain complex III and complex IV deficiency, and the other isolated complex I deficiency.
One of the patients presented with impaired motor skills, hypotonia, epileptic seizures and comprehension and language delays. Suspicion of reduced levels of serotonin metabolites in cerebrospinal fluid led to initiation of oral hydroxytryptophan at a daily dose of 200 mg compounded in the pharmacy department. At present, although still experiencing frequent falls, the patient exhibits improvements in motor skills, comprehension and expressive language.
The other patient had onset at age 3 months with anorexia, neurosensory hearing loss, atrial septal defect and bilateral ventriculomegaly. The relevant family history was the death within 6 months of birth of 5 siblings of the father for unknown causes. The patient underwent gastrostomy with placement of a feeding tube due to poor oral tolerance. At present, she is receiving oral riboflavin at a dose of 60 mg every 12 h and sodium succinate at 1 g every 12 h, both compounded in the pharmacy. She exhibits severe psychomotor and cognitive impairment, with absence of verbal language and bowel and bladder control. The patient wears restraints to keep her from hurting herself. She also has chronic constipation, recurrent urinary tract infections and several dental health problems.
A boy aged 6 years started exhibiting developmental impairment at age 1 year characterised by hypotonia and sucking, swallowing and coordination problems that precluded adequate nutrition. In 2015, the patient underwent genetic testing in the referral hospital for diagnosis of psychomotor delay and dystonic tetraparesis of unknown aetiology, with suspicion of a mitochondrial defect or neurotransmitter deficiency. Cerebrospinal fluid analysis revealed abnormal levels of 5-hydroxytriptophan (45 nmol/L; normal range, 0–26), 5-hydroxyindoleacetic acid (11 nmol/L; normal range, 170–440) and homovanillic acid (282 nmol/L; normal range, 344–466), prompting initiation of treatment with carbidopa/levodopa and 5-hydroxytryptophan. The clinical presentation consisted of spasticity in the lower extremities, balance disorder, hypersalivation and occasional dysphagia. The patient has exhibited improvements in motor skills, as he is currently able to walk independently with the aid of a walker, as well as in communication, language and manipulation skills. He has adapted well to school. Due to development of constipation, it has not been possible to reach the full dose of hydroxytryptophan.
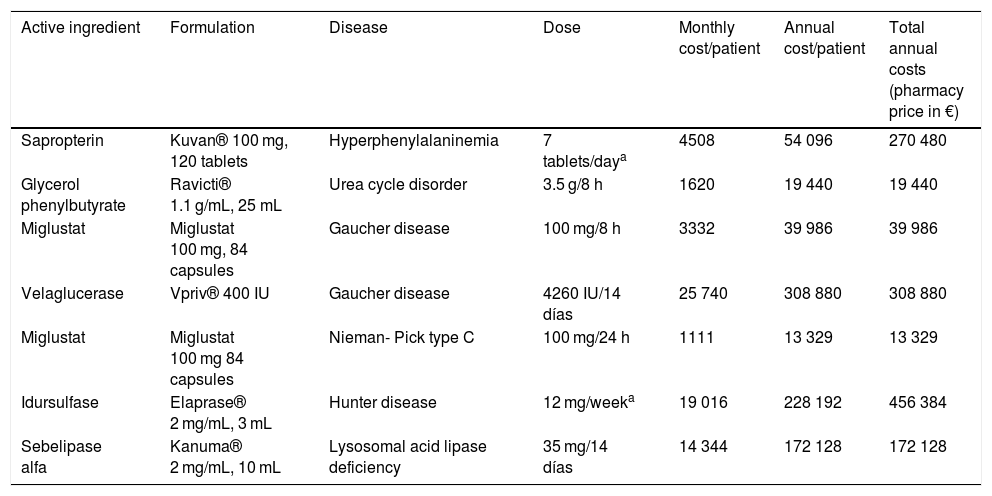
Economic analysisTable 3 presents the results of the economic analysis of the use of orphan drugs for treatment of CEMs.
Cost of orphan drugs used to treat congenital errors of metabolism.
| Active ingredient | Formulation | Disease | Dose | Monthly cost/patient | Annual cost/patient | Total annual costs (pharmacy price in €) |
|---|---|---|---|---|---|---|
| Sapropterin | Kuvan® 100 mg, 120 tablets | Hyperphenylalaninemia | 7 tablets/daya | 4508 | 54 096 | 270 480 |
| Glycerol phenylbutyrate | Ravicti® 1.1 g/mL, 25 mL | Urea cycle disorder | 3.5 g/8 h | 1620 | 19 440 | 19 440 |
| Miglustat | Miglustat 100 mg, 84 capsules | Gaucher disease | 100 mg/8 h | 3332 | 39 986 | 39 986 |
| Velaglucerase | Vpriv® 400 IU | Gaucher disease | 4260 IU/14 días | 25 740 | 308 880 | 308 880 |
| Miglustat | Miglustat 100 mg 84 capsules | Nieman- Pick type C | 100 mg/24 h | 1111 | 13 329 | 13 329 |
| Idursulfase | Elaprase® 2 mg/mL, 3 mL | Hunter disease | 12 mg/weeka | 19 016 | 228 192 | 456 384 |
| Sebelipase alfa | Kanuma® 2 mg/mL, 10 mL | Lysosomal acid lipase deficiency | 35 mg/14 días | 14 344 | 172 128 | 172 128 |
The use of orphan drugs for CEMs generate total annual costs of 1 290 674 euro, corresponding to approximately 3.0% of the pharmacy budget of our hospital (using the total expenditure of 2018 as reference). The treatments that are most costly per patient are, in decreasing order, velaglucerase, idursulfase and sebelipase alfa, all of which are delivered intravenously. In terms of total costs, the highest expenditure corresponds to idursulfase, followed by velaglucerase and sapropterin, the treatments that have the highest impact on the budget. The most inexpensive drugs are miglustat and glycerol phenylbutyrate.
DiscussionThe consumption of orphan drugs in Spain at the hospital level amounted to 8.4% of the hospital-based health care expenditures in Spain in 2016,22 which was consistent with other European countries.23 Orphan drugs used for treatment of CEMs corresponded to 1.3% of the total pharmacy expenditure of the hospital, so the overall expenditure in orphan drugs for treatment of any condition is likely to exceed the European average.
Taking into account the reported prevalence of CEMs and the total catchment population of the hospital of approximately 300 000 individuals, we could estimate the expected prevalence in our study.24 The number of patients with Gaucher disease, Hunter syndrome and LALD was consistent with the expected frequency. The frequency of phenylketonuria, urea cycle disorders and Niemann-Pick disease was lesser than predicted. In the latter two, the lower prevalence may be due to underdiagnosis, due to early mortality or because these patients may not always need an orphan drug (2 of the 6 urea cycle disorders are treated exclusively with arginine).14
Clinical trials of sapropterin found mean levels of phenylalanine of 607 nmol/mL after 6 months of treatment.5 Our patients had levels close to the treatment threshold. This could be due to the short duration of the trial compared to our study, in which patients had been in treatment for years.
The efficacy of glycerol phenylbutyrate has been assessed in 2 studies, which found that blood ammonia levels stayed in the10−51 µg/dL range.6 The decreased effectiveness observed in our study could be due to the patient initiating treatment in the advanced stages of disease, whereas patients recruited to the trial could not have any clinical manifestations of hyperammonaemia. Among other adverse effects, our patient developed insomnia, which was not mentioned in the summary of product characteristics.6
Miglustat was authorised for treatment of Gaucher disease after a clinical trial in 28 patients. The trial found a decrease in hepatomegaly in addition to a mean increase in the platelet count of 22.2 × 109/L and in the haemoglobin concentration of 0.95 g/dL.15 The effectiveness observed in our study was slightly lower, probably due to the performance of splenectomy in the patient, a treatment that is no longer recommended today and that delayed initiation of pharmacotherapy.15
The efficacy of velaglucerase was assessed in 4 studies that used normalization of the haemoglobin concentration and platelet count as endpoints. The extension study found normalization of the liver and spleen volumes.7 Treatment in our patient achieved all the therapeutic goals, with outcomes similar to those reported in the studies.
The efficacy of miglustat for treatment of Niemann-Pick disease was assessed in a prospective clinical trial and a retrospective review, which found evidence of amelioration of clinically significant neurologic manifestations.8 This approach to assessing effectiveness is subjective, and objective measures should be established for its assessment.
Two clinical trials have demonstrated the safety and efficacy of idursulfase in patients aged more than 5 years. The efficacy was assessed with a composite endpoint based on the 6MWT and the FVC.9 We did not perform spirometry in our patients because its results in children are not reliable. The 6MWT was only performed in one patient, whose result was better compared to the clinical trial (43.3 m). It would be useful to assess whether the 6MWT was performed under the same conditions as in the clinical trial. The secondary endpoints were the urine levels of GAGs, which normalised in 50% of patients, and the control of visceromegaly.25 In our patients, we observed a significant reduction in GAG levels at the beginning of treatment, but they soon plateaued above the upper limit of normal. Both patients achieved normalization of visceral organ volume. In terms of safety, one of the patients experienced acral coldness, which was not described in the summary of product characteristics.9
Sebelipase alpha was found to be effective in a multicentre study. In our study, the reduction in triglyceride and low-density lipoprotein relative to baseline was greater compared to the previous study, which had found reductions of 25% and 28%, respectively. The increase in HDL was inferior to the 20% increase observed in the trial. The clinical trial included the normalization of ALT levels and their reduction relative to baseline (which was of 53%) as endpoints.10 In our patient, treatment did not achieve normalization, although levels neared the upper limit of normal, but there was an estimated reduction of 58%. The effectiveness in our study seemed superior compared to the efficacy found in clinical trials.10 Possible explanations include the baseline lipid levels of our patient being better than those of trial participants and that the trial lasted 20 weeks, compared to the year and a half of follow-up in our study.
We assessed the effectiveness and safety of compounded preparations used for treatment of mitochondrial respiratory chain defects with subjective criteria. It would be helpful to perform research assessing the clinical criteria that may be used in measuring the effectiveness of compounded formulations for treatment of different diseases for the purpose of developing guidelines or protocols.
There is no question that clinical trials to date have provided little safety and efficacy data, in addition to the potential limitations in their design: small sample size, single treatment arm, absence of active controls, patients of heterogeneous demographic characteristics, etc. Our study also has limitations: it was conducted in a single centre in a small sample that did not guarantee representation of the population. There is also a risk of underestimating frequencies, as electronic health record forms are not designed to serve as data sources. Performance of multicentre studies with larger samples allowing separate analysis of different CEMs would be necessary to obtain robust data.
It is fair to conclude that the effectiveness of orphan drugs for treatment of CEMs is heterogeneous. Some, such as glycerol phenylbutyrate or idursulfase, achieve mild improvement, while others achieve clinical stabilization or even the absence of symptoms, for instance in Gaucher disease.
Overall, patients tolerated these drugs well, with the exception of one patient that developed paroxysmal supraventricular tachycardia secondary to treatment with idursulfase.
Due to the uncertainty regarding the clinical evidence on the efficacy of orphan drugs and the high costs associated with their use, the cost-effectiveness of these drugs remains a controversial subject.
FundingThis research did not receive any external funding.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Caso-González A, Núñez-Rodríguez J, Nebot-Villacampa M-J, González-Pérez Y, Marín-Gorricho R, Leralta-González C, et al. Experiencia clínica con medicamentos huérfanos para enfermedades raras metabólicas. An Pediatr (Barc). 2022;96:8–16.
