





The screening programme of congenital hypothyroidism (CH) is probably one of the best achievements in paediatrics. Thyroid hormones are essential for brain development and brain maturation that continue through the neonatal period. Hypothyroidism that begins in the first months of life causes irreversible damage to the central nervous system, and is one of the most frequent and preventable causes of mental retardation. As children with congenital hypothyroidism are born with a normal appearance, analytical studies are required to immediately start the appropriate therapy.
This article analyses the aims, diagnostic procedures, tests required, aetiology, and differential diagnosis in this disorder. Especially relevant is to perform frequent monitoring to ensure dose adjustments of L-Thyroxine therapy, avoiding infra- or supra-dosing that negatively affects neurosensory functions. Re-evaluation of the aetiology permanent vs transient hypothyroidism is always recommended after 3 years of chronological age.
The relevance of this screening programme should be widely discussed in paediatrics. The main objective is to avoid cerebral damage in these patients, and has been highly successful and economically beneficial.
Other aspects are required to optimise patient outcomes, to perform all the controls according to the recommendations and to include, in the near future, the diagnosis of central hypothyroidism. Implementation of this programme is necessary to progress in accordance with current scientific knowledge.
El Programa de cribado o detección precoz del hipotiroidismo congénito (HC) es uno de los mayores avances logrados en Pediatría. Las hormonas tiroideas son imprescindibles para el desarrollo y la maduración cerebral, que continúan en la etapa neonatal. El hipotiroidismo de comienzo en los primeros meses de vida origina lesiones irreversibles en el sistema nervioso central y es una de las causas más frecuentes y evitables de retraso mental. El diagnóstico clínico es tardío, por lo que requiere estudio analítico para poder efectuar el tratamiento adecuado.
Este artículo actualiza los objetivos, los procedimientos diagnósticos, las pruebas imprescindibles y complementarias requeridas, la etiología y los diagnósticos diferenciales en esta patología. Con especial énfasis en los requerimientos de los centros de seguimiento para protocolizar los resultados del tratamiento con L-tiroxina administrada de forma inmediata al diagnóstico y a las dosis que eviten fases de infra o supradosificación que pueden alterar diversos aspectos del desarrollo cognitivo. La revaluación de etiología permanente vs. transitoria se recomienda siempre después de los 3 años de edad.
La relevancia de este programa precisa su difusión a todas las áreas de pediatría. El objetivo principal, evitar el daño cerebral en estos pacientes, se ha logrado y es además altamente beneficioso desde el punto de vista económico.
Otros aspectos para optimizar los resultados cognitivos con todos los controles periódicos necesarios y lograr la inclusión del diagnóstico del HC central, precisan implementar los recursos de los centros de seguimiento y continuar avanzando según los conocimientos actuales.
Hypothyroidism is characterised by the clinical and laboratory features that result from the decreased biological activity of thyroid hormones at the tissue level. In most cases, it is associated with decreased serum levels of thyroid hormones with elevation of thyroid stimulating hormone (TSH) levels.
Congenital hypothyroidism (CHT) and neonatal hypothyroidism comprise a heterogeneous group of thyroid hormone abnormalities leading to reduced thyroid function that can be detected as early as the neonatal period.1,2
Thyroid hormones are essential to achieve normal growth and brain maturation. Hypothyroidism with onset in the early months of life may cause irreversible lesions in the central nervous system. Congenital and neonatal hypothyroidism must be diagnosed and treated on an urgent basis, as they are a frequent and preventable cause of intellectual disability.3–6
The symptoms are nonspecific and progress in direct correlation to the duration and the severity of hypothyroidism. In the first month of life, when treatment should be initiated, only 5% of affected individuals could be diagnosed based on symptoms alone.
The first programme for neonatal screening of CHT was implemented in Quebec in 1974.1 At present, most developed countries offer routine screening, but this is not the case in developing countries.7
Screening programmes have evinced that CHT is very frequent and have prevented, in most cases, the brain damage and secondary permanent intellectual disability that had been observed in these children in the past.
The prevalence of primary CHT (which is due to an absent or defective thyroid) reported in international case series is of 1 per 2000 live births, which is consistent with the figures reported for Spain.7,8
Objective of the neonatal screening programme for congenital hypothyroidismThe Programme for the Early Detection of Congenital Hypothyroidism, a public health and preventive medicine priority, is part of the neonatal screening programme.
Its main goal is the detection and treatment of severe and permanent CHT. Detection of mild permanent CHT or transient CHT is a secondary objective of screening.
In all of these forms of CHT, early detection prevents neurologic damage and reduces the morbidity, mortality and potential disabilities associated with the disease.
It is important to emphasise that neonatal screening tests are not diagnostic procedures. Patients with a positive result of screening require additional diagnostic evaluation, which requires the support of clinicians specialised in the diagnosis and treatment of the disease (clinical follow-up centres [CFUCs]). Clinical follow-up centres should meet certain conditions (Tables 1 and 2). Their purpose is to confirm or rule out the diagnosis of CHT, and promptly initiate treatment if applicable.8,9
Objectives of clinical follow-up centres for congenital hypothyroidism.
| Confirming the CHT diagnosis through history-taking, physical examination and the necessary diagnostic tests (blood and imaging tests) with immediate turnaround of results. |
| Informing and reassuring the family; explaining the benefits of early detection |
| Urgent initiation of treatment (levothyroxine) with optimisation of treatment during follow-up visits until thyroid function normalises |
| Achieving a normal neurologic and psychomotor development and an intellectual quotient within the normal range while preventing comorbidities |
| Diagnosing the aetiology (permanent vs transient CHT) |
| Diagnosing other potentially associated congenital anomalies |
| Ongoing bidirectional information sharing with the diagnosing facility to assess the outcomes and effectiveness of the programme |
| Establishing a direct relationship with the patient's paediatrician to coordinate care |
Requirements for clinical follow-up centres for congenital hypothyroidism.
| Paediatric endocrinologists with experience in CHT and full-time dedication |
| Department of Biochemistry with rapid testing capabilities for diagnostic confirmation and follow-up evaluation |
| Department of Nuclear Medicine (initial urgent thyroid scintigraphy) |
| Department of Paediatric Radiology (thyroid ultrasound, bone age study) |
| Department of Psychology for evaluation |
| Inpatient and neonatal care services |
| Relationship with the patient's paediatrician |
| Direct communication with the diagnosing institution to assess the outcomes and efficacy of the programme |
| Periodic communication with the Subdirectorate General of Health Promotion and Prevention of the Department of Health or the institution overseeing the programme |
| Expert Committee meetings to assess and improve programme outcomes |
Neonatal screening should not be understood as a mere laboratory procedure, but as a multidisciplinary activity whose coordination with the health care system is essential to ensure its effectiveness and efficiency.
Procedure for neonatal screening for congenital hypothyroidismMeasurement of TSHThe collection of samples is scheduled so that coverage of 100% of newborns is ensured, along with treatment of 100% of the detected cases.
Screening for early detection of primary congenital hypothyroidism is performed by measurement of the levels of TSH at 48h post birth to avoid the initial physiological elevation of this hormone after birth.
A capillary blood sample is obtained from the newborn by heel puncture and collected on standard filter paper; this procedure needs to be performed by specially trained health care staff to optimise yield.8
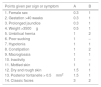
The serum levels of TSH are measured by immunofluorescence assay (DELFIA®, Laboratorios Perkinelmer). The cut-off point above which CHT is suspected has been established at 7–10IU/mL. When the TSH value exceeds the established threshold, the total thyroxine level (tT4) is measured as well.
At present, all centres that screen for CHT in Spain measure the levels of TSH, while the levels of both TSH and tT4 are only measured simultaneously in the initial test in a few autonomous communities (Basque Country, Navarre and Cantabria).
Central hypothyroidism (secondary or tertiary) is not detected by screening programmes that only measure TSH.10,11
Confirmation of congenital hypothyroidismIn case of a positive screen result, the facility that did the screening urgently contacts the patient for referral to the corresponding CFUC, where testing will be performed for confirmation of hypothyroidism and to establish the aetiology of the abnormal thyroid function, without delaying initiation of treatment.12–17
The aetiology of congenital hypothyroidism is multifactorial. In 95% of cases the patient has primary hypothyroidism (abnormalities in the thyroid gland), while central hypothyroidism is less frequent.
Congenital and neonatal hypothyroidism are present from birth. Depending on the aetiology, the hypothyroidism may be permanent and require lifelong treatment, or it may be transient and resolve spontaneously when the underlying cause disappears (e.g. excessive concentration of iodine or the presence of maternal antithyroid antibodies). Notwithstanding, every patient requires treatment to normalise thyroid function during the period of brain development, and treatment is inefficient if it starts later (existence of a “window of opportunity”).4
The most frequent cause of primary CHT is thyroid dysgenesis (85%–90%), in most cases thyroid ectopia (60%–65%), followed by thyroid agenesis or athyreosis (35%–40%). Different forms of thyroid dyshormonogenesis (Table 3) account for 10% of cases. Specific mutations in the transcription termination factor 2 gene (TTF2) and genes encoding other thyroid transcription factors have been identified in some patients with thyroid dysgenesis.
Aetiology of congenital hypothyroidism. Thyroid dyshormonogenesis.
| 1. Defects in iodine trapping or transport |
| 2. Defects in organification |
| Mutation in thyroid peroxidase gene |
| Defects in hydrogen peroxide generation |
| Iodine acceptor abnormalities |
| Pendred syndrome |
| 3. Defective iodotyrosine coupling |
| Hollander syndrome |
| 4. Iodotyrosine dehalogenase deficiency |
| 5. Abnormal iodoproteins |
| 6. Abnormalities in thyroglobulin synthesis |
| Mutation in thyroglobulin gene |
| Decreased thyroglobulin mRNA levels |
| Truncated thyroglobulin protein |
| Sialic-acid-deficient thyroglobulin |
| 7. Insensitivity to TSH |
In some cases, the aetiology of hypothyroidism cannot be determined. Numerous genes may be involved in the multifactorial aetiology of primary congenital hypothyroidism.17–24
Reporting test resultsThis is a key step in the diagnosis and treatment of CHT. The purpose is to help parents understand the benefits of early diagnosis in the prevention of brain damage, teach them how to correctly administer medication, and promote adherence to treatment and to follow-up care for the duration of CHT (months, years, or life). The initial results should be conveyed by experienced staff, ensuring that parents fully understand the information.
Management at the clinical follow-up centre for congenital hypothyroidismFig. 1 presents the protocol for management following a positive result of neonatal screening.
A detailed personal and family history is taken in every case, with particular emphasis on potential consumption of pharmacological agents or iodine-containing substances, family history of thyroid disease (especially in the mother) and of symptoms of underactive thyroid.
The patient also undergoes a clinical evaluation to identify the signs and symptoms of hypothyroidism (Table 4).
Clinical signs and symptoms of congenital hypothyroidism. Congenital hypothyroidism index.
| Points given per sign or symptom | A | B |
|---|---|---|
| 1. Female sex | 0.3 | 1 |
| 2. Gestation >40 weeks | 0.3 | 1 |
| 3. Prolonged jaundice | 0.3 | 1 |
| 4. Weight >3500g | 0.5 | 1 |
| 5. Umbilical hernia | 1 | 2 |
| 6. Poor sucking | 1 | – |
| 7. Hypotonia | 1 | 1 |
| 8. Constipation | 1 | 2 |
| 9. Macroglossia | 1 | 1 |
| 10. Inactivity | 1 | – |
| 11. Mottled skin | 1 | 1 |
| 12. Dry and rough skin | 1.5 | 1 |
| 13. Posterior fontanelle > 0.5mm2 | 1.5 | 1 |
| 14. Classic facies | 3 | 2 |
Clinical score suggestive of congenital hypothyroidism: >4 points.
Source: Letarte A. et al., 1980.12
The results of the dried blood spot test are confirmed by venous blood tests (TSH, free thyroxine [fT4]). In primary hypothyroidism, elevated TSH levels are associated with reduced levels of fT4. Thyroglobulin levels are measured to assess for the presence of the thyroid gland. The presence of antithyroid antibodies supports the diagnosis of transient autoimmune thyroiditis due to hormone blocking. Elevation of iodine levels in the urine (>200μg/L) suggests excessive exposure to iodine with blocking of thyroid receptors.
Imaging tests, such as thyroid ultrasound and scintigraphy, are very useful in determining the aetiology of CHT. They allow the definitive diagnosis of dysgenesis, while detection of an eutopic thyroid gland with normal tracer uptake may suggest transient CHT.
A thyroid ultrasound examination can verify the presence of thyroid tissue in the neck. It can differentiate between thyroid dysgenesis (agenesis or ectopia) and dyshormonogenesis, which manifests with presence of the thyroid gland in the normal location. However, the interpretation of thyroid ultrasound findings in the newborn requires a high degree of specialisation.
Thyroid scintigraphy with sodium iodide I 131 or technetium Tc 99m can be used to determine the position and size of the thyroid gland and establish a definitive diagnosis of thyroid agenesis, ectopia, hemiagenesis or hypoplasia. If the thyroid cannot be visualised with scintigraphy in a patient with elevated thyroglobulin levels, it may be helpful to perform an ultrasound scan, as this may be due to the presence of thyroid-blocking antibodies (maternal autoimmune thyroid disease), a defect in iodide trapping in cases of dyshormonogenesis (NIS mutation), a genetic change in the TSH-β gene or inactivating mutations of the TSH receptor gene. In cases of partial block of thyroid hormonogenesis, the thyroid may be eutopic and of normal size.
Other tests (not essential to the initial diagnosis)Auditory evoked potentials: the relationship between hypothyroidism and hearing loss is well established. Some forms of thyroid dyshormonogenesis (Pendred syndrome, Hollander syndrome) should be suspected if there is a family history of hearing loss.
Congenital malformations may be more frequent in patients with CHT compared to the general population, especially congenital heart defects, and therefore, performance of a cardiologic evaluation is recommended in these patients. An evaluation by a paediatric neurologist at the time of diagnosis is also very useful to detect potential comorbidities associated with psychomotor retardation.
Differential diagnosisIf CHT is detected in a newborn through screening and later confirmed by the thyroid function study conducted in the CFUC, differential diagnoses need not be considered and treatment must not be delayed under any circumstances to attempt to determine the exact aetiology of hypothyroidism. In many cases, the definitive diagnosis is established in the re-evaluation performed at 3 years of chronological age, while in other cases, especially those of transient CHT, the aetiology may not ever be established.
Paediatricians have to evaluate newborns from countries where screening is not performed to verify normal thyroid function.
There are special situations where performance of repeat TSH measurements at 2 and 4 weeks of life is recommended due to the potential for delayed elevation of TSH or the need to measure both T4 and TSH (Table 5).25–28
Special situations in the early detection of congenital hypothyroidism.
| Newborns with gestational age ≤30 weeks |
| Birth weight ≤1500g |
| Use of iodine antiseptic products during delivery or in the care of the newborn (caesarean section, major surgery) |
| Performance of radiologic tests with iodinated contrast media |
| Silastic catheter insertion, cardiac catheterization |
| Down syndrome |
| Multiple birth, especially twins of the same sex |
| (potential for foeto-foetal transfusion syndrome) |
| Newborns admitted to intensive care unit |
| Post-transfusion samples |
Source: Rodríguez Sánchez et al.15
In congenital and neonatal hypothyroidism, the degree of brain damage is directly associated with the time elapsed between onset of hypothyroidism and initiation of treatment. Thus, treatment should be initiated as soon as possible, preferably before age 15 days. Treatment should start the day the diagnosis is made, and its initiation should not be delayed pending performance of additional diagnostic tests.
The first-line drug is synthetic levothyroxine via the oral route, administered 30min before a feeding once every 24h at a dose of 10–15μg/kg/day.29–33
Generic formulations of levothyroxine do not exhibit the same bioavailability.34 Liquid compounded formulations of levothyroxine are not stable. The absorption of levothyroxine is hindered by ingestion of foods or products containing soy, iron, calcium, aluminium hydroxide, omeprazole, fibre, sucralfate or bile acid sequestrants.
Once treatment with levothyroxine starts, the patient must undergo frequent clinical and laboratory follow-up evaluations to optimise treatment. We recommend a second visit 2 days after diagnosis to confirm acceptance of the diagnosis and prescribed treatment, a third evaluation at 15 days with measurement of TSH and fT4 levels, then monthly visits in the first 6 months of life, followed by visits every 2 months until age 1 year, and visits every 3–4 months until the patient is re-evaluated at age 3 years.
Each follow-up visit should include a full physical examination and measurement of fT4 and TSH levels. The aim of treatment is to quickly normalise fT4 and TSH levels. Free thyroxine levels must remain in the upper range of normal to avoid subclinical hypo- and hyperthyroidism.35 The levels of TSH and fT4 must be measured 4 weeks after any dosage change.
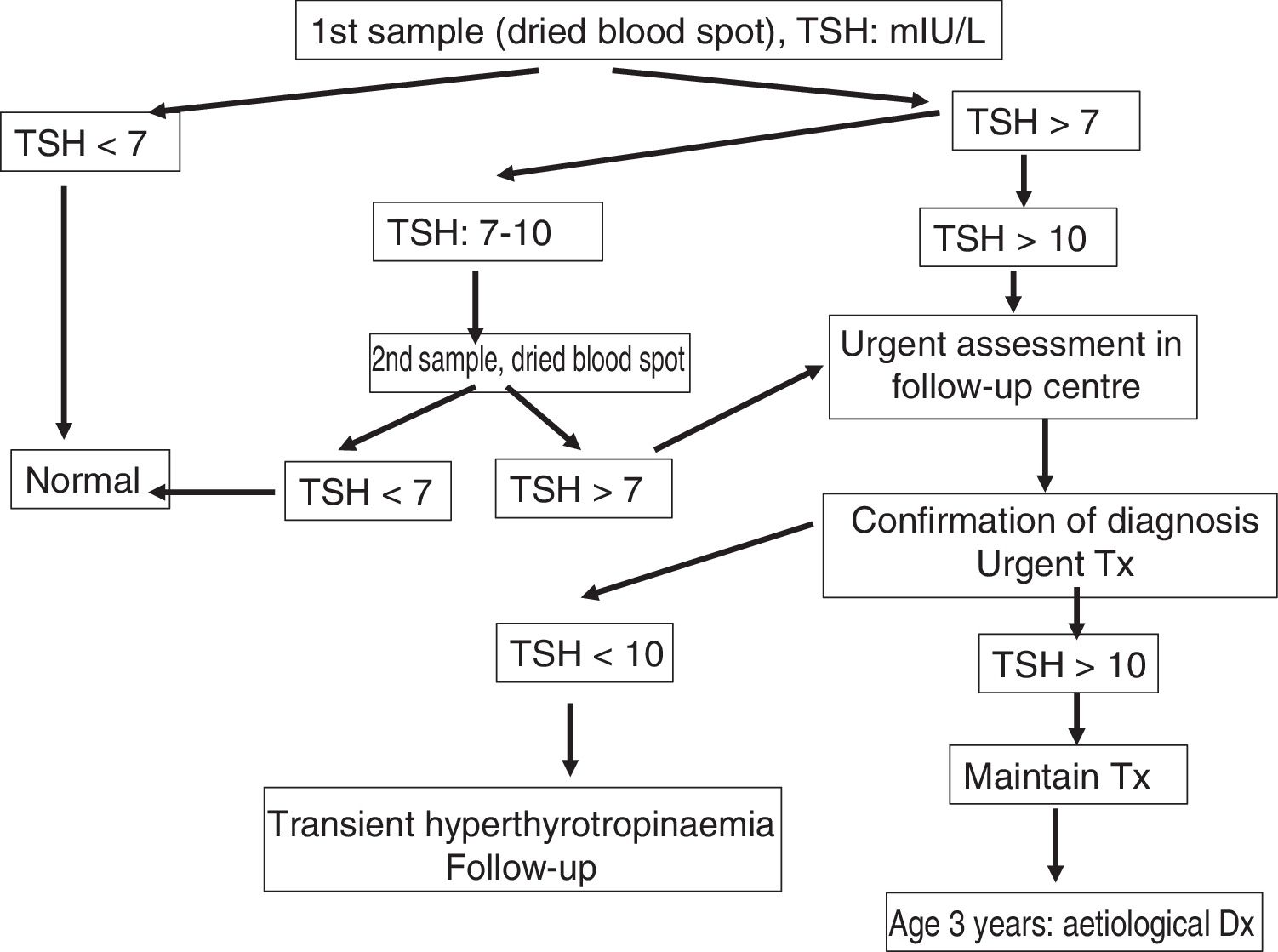
Definitive diagnosis of congenital hypothyroidismThe treatment of children identified through screening programmes for early detection of CHT must be maintained without interruption through age 3 years to ensure normal thyroid function until brain growth is complete, whether the hypothyroidism is transient or permanent. At age 3 years, patients without a definitive diagnosis may undergo re-evaluation (Fig. 2).
Children with an initial diagnosis of thyroid agenesis or ectopic thyroid, which are both permanent, do not require re-evaluation. The diagnosis ought to be re-evaluated in children with CHT and a normally positioned thyroid at the time of initial diagnosis, and in children with CHT of unknown aetiology.36,37 Re-evaluation should also be contemplated in patients in whom the diagnosis was not confirmed (by scintigraphy) at the time treatment was initiated. There are 2 possible approaches to the diagnostic re-evaluation, which should always be performed after the patient reaches age 3 years.
To determine whether the hypothyroidism is permanent or transient, it is sufficient to half the current dose of levothyroxine and measure the levels of TSH and fT4 one month after: if the TSH level is 10mIU/L or higher, the hypothyroidism is considered permanent and the patient will resume treatment with levothyroxine at the dose that achieved normal thyroid function. In patients with a very high initial level of TSH and an eutopic thyroid gland, discontinuing levothyroxine for just 2 weeks should be enough to produce elevation of TSH.
If the aim is to make a definitive diagnosis, treatment with levothyroxine should be suspended for 1 month, followed by measurement of levels of fT4, TSH, thyroglobulin and antithyroid antibodies (antithyroglobulin or anti-thyroid peroxidase). If it was not performed before initiation of treatment, thyroid scintigraphy with technetium 99 can be used to diagnose an ectopic thyroid or possibly thyroid agenesis, which would be confirmed with a thyroid ultrasound scan. In patients with permanent CHT with eutopic thyroid gland, thyroid scintigraphy with sodium iodide I 131 allows performance of a perchlorate discharge test for assessment of dyshormonogenesis. Molecular genetic testing is very useful in the diagnosis of CHT.
The following findings are suggestive of permanent CHT in patients with an eutopic thyroid gland: history of goitre or primary congenital hypothyroidism with eutopic thyroid gland in first-degree relative, thyroid hypoplasia at the time of initial diagnosis, need for high doses of levothyroxine (>2μg/kg/day) during the follow-up, thyroid hyperplasia at the time of re-evaluation.
Table 3 summarises the aetiological classification of CHT based on the results of the diagnostic re-evaluation. If a permanent aetiology is confirmed, the family needs to be informed of the need to maintain treatment with levothyroxine for life with the required dose adjustments. In case of transient hypothyroidism, treatment is discontinued.
PrognosisEarly detection of CHT through neonatal screening prevents the intellectual disability formerly observed in these children when the diagnosis was clinical and delayed. Patients with CHT that receive early and adequate treatment have normal intellectual quotients.17,18,36,38,39
In some cases, mild brain impairment occurs despite early initiation of treatment, resulting in behavioural problems, and impairment in language comprehension, fine motor skills and visual-perceptual motor skills, usually with little impact on the ability to carry out a normal life. These abnormalities have been associated with the age at initiation of treatment (greater than 15–21 days), the dose of levothyroxine (usually lower than recommended), the initial severity of hypothyroidism and fT4 levels below or above the recommended range during the follow-up. Excessive levels of fT4 in the early months of life may be associated to inattention in subsequent years.35
Discontinuation of treatment before age 3 years without medical supervision or subsequent follow-up can have dire effects on neurologic development.
Follow-up in these patients is important not only to achieve healthy maturation, but also optimal growth and development. In countries where there are no CFUCs for patients with CHT, the reported proportion of patients that discontinue treatment prematurely ranges between 25% and 30%.37,40
Transition to adult careChildren with CHT should be followed up in a CFUC through puberty and until reaching their final height.16,17 Thereon, follow-up visits should be scheduled every 6–12 months, or more frequently in case of poor adherence. The purpose of followup is to monitor thyroid function to prevent subclinical hypo- or hyperthyroidism and the associated cardiovascular problems, excess weight and bone mineralisation defects in adulthood.
Only 70% of adults with CHT adhere to treatment correctly. This is particularly alarming in the case of women who are or wish to become pregnant, whose thyroid hormone requirements are greater. Optimal treatment of maternal hypothyroidism is essential to achieve a normal pregnancy with healthy neurocognitive development in the child. Monitoring of treatment is recommended throughout childhood and adulthood. Educational strategies need to be developed and implemented to improve adherence to treatment, especially during the transition to adult care.
Unresolved problems in the early detection of congenital hypothyroidismEven if the results of screening are normal, if there is clinical suspicion of hypothyroidism the paediatrician should request urgent measurement of serum fT4 and TSH levels, as the elevation of TSH may occur later in some patients. Other infrequent causes, such as consumptive hypothyroidism in the postnatal period secondary to overexpression of type 3 deiodinase in patients with large haemangiomas, will not be detected in screening tests due to normal levels of TSH and T4 in the early stages.27
The screening protocol that measures TSH alone cannot identify cases of central CHT.10,11 The incidence of central CHT, whether isolated or associated with hypopituitarism, is higher (1 per 16,000 to 20,000 live births) than previously reported (1 per 100,000), so expanded screening with simultaneous measurement of TSH and tT4 levels is recommended. For now, we advocate for measurement of T4 levels in the newborn screening sample, as this would prevent delayed diagnosis in these patients, in many of whom symptoms may be either absent or nonspecific.
Abnormalities in TSH-α and TSH-β may manifest with a late elevation of TSH. Changes in the monocarboxylate transporter 8 (MCT8) gene should be suspected in patients with neurologic manifestations in the first year of life and initially manifest with normal thyroid function.16,17
ConclusionsThe Programme for the Early Detection of CHT constitutes one of the greatest advances in child health and preventive medicine of the XX century. The identification of these patients prevents the suffering involved in living with permanent brain damage in both the individual and the family. From an economic perspective, these programmes offer a high yield to society, yet only 25% of the global population benefits from them.
Further cost-benefit analyses are required to establish a consensus regarding the need to measure the levels of only TSH, only T4 or both to allow detection of central hypothyroidism too, and whether the cut-off point for TSH should be lowered to 5mIU/L to allow detection of milder forms of hypothyroidism while avoiding an increase in the false-positive rate.
For all the above reasons, in upcoming years the necessary infrastructure should be developed to provide these programmes with the capability to apply current scientific knowledge, and their positive outcomes disseminated effectively in health care and social forums.
Conflicts of interestThe authors have no conflicts of interest to declare.
Caimari Jaume, María, Hospital Universitario Son Espases, Balearic Islands; Casano Sancho, Paula Hospital Sant Joan de Déu, Barcelona; Grau Bolado, Gema, Hospital Cruces, Bizkaia; Muñoz Calvo, M. Teresa, Hospital Infantil Niño Jesús, Madrid; Rial Rodríguez, José Manuel, Hospital Universitario Nuestra Señora de Candelaria, Santa Cruz de Tenerife, and Temboury Molina, Carmen, Hospital del Sureste, Madrid.
The following is the supplementary data to this article:

Please cite this article as: Rodríguez Sánchez A, Chueca Guindulain MJ, Alija Merillas M, Ares Segura S, Moreno Navarro JC, Rodríguez Arnao MD, et al. Diagnóstico y seguimiento de los pacientes con hipotiroidismo congénito diagnosticados por cribado neonatal. An Pediatr (Barc). 2019;90:250.
Appendix 1 lists the members of the Thyroid Working Group of the Sociedad Española de Endocrinología Pediátrica (SEEP).
To produce this work, we used the Appraisal of Guidelines for Research and Evaluation (AGREE) Instrument, Spanish version (www.agreecollaboration.org).

Anales de Pediatría (English Edition) follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals