



Smith-Lemli-Opitz syndrome (SLOS) is an inborn error of cholesterol metabolism due to a deficiency of the enzyme 3 beta-hydroxysterol-delta 7-reductase, which usually results in elevation of 7-dehydrocholesterol levels and decreased cholesterol levels in blood and other tissues during foetal development and after birth. The syndrome is caused by biallelic pathogenic changes in the DHCR7 gene. The classical phenotype is characterised by prenatal and postnatal growth restriction, microcephaly, multiple major and minor malformations (mainly cardiac defects, cleft palate and ambiguous genitalia) and moderate-to-severe intellectual disability.1 For reasons that remain unknown, its expressivity varies widely,2 ranging from exceptional mild cases with normal cognitive function3 and severe cases manifesting with central nervous system anomalies such as holoprosencephaly.4
We present 4 cases diagnosed in 2 unrelated families that represent mild and severe forms of the disease.
Family 1. Girl aged 8 years born to healthy, nonconsanguineous parents, evaluated at birth due to multiple congenital anomalies: microcephaly, dysmorphic facies, punctiform cataracts, soft palate cleft, second-toe and third-toe syndactyly and hypotonia with abnormal suck. She received a clinical diagnosis of SLOS (classic phenotype), and measurement of sterol levels confirmed the elevation of 7-dehydrocholesterol (436.3 mmol/L) and decreased total cholesterol levels (27 mg/dL). Treatment with cholesterol was initiated immediately. Sequencing of the DHCR7 gene confirmed the suspected diagnosis through the identification of the pathogenic variants c.906C>G/c.964-1G>C.
During the follow-up, the patient developed severe gastro-oesophageal reflux, severe global developmental delay (absence of walking and language development) and epilepsy with onset at 9 months and poor control of seizures despite treatment with levetiracetam, lamotrigine, zonisamide and perampanel; moderate bilateral neurosensory hearing loss; recurrent respiratory tract and ear infections and obesity.
The relevant family history consisted of termination of pregnancy by the parents in week 20 of a genetically male foetus (46, XY) with female sexual characteristics and holoprosencephaly on ultrasound. After SLOS was diagnosed in the patient, genetic testing of amniocytes in this pregnancy confirmed that the foetus was also affected.
Family 2. Man aged 22 years, born to healthy, nonconsanguineous parents, evaluated at age 12 years for growth restriction of prenatal onset, microcephaly, cleft palate, abnormal facies (long face, high nose bridge, wide nostrils and mild downward slant of palpebral fissures), 2nd-3rd toe syndactyly, and short index finger attached low in the hand, as can be seen in Fig. 1. The patient had a brother aged 18 years with prenatal and postnatal growth restriction, bifid uvula, physically resembling the patient and with mild impairment in social skills. Both patients exhibited normal psychomotor development. Neither had cardiac or genitourinary anomalies. The initial diagnostic tests were as follows: the results of serum cholesterol measurement (145-170 mg/dL), karyotyping (46,XY), MLPA 22q11 sequencing and cranial magnetic resonance imaging were normal, and maternal phenylketonuria was ruled out. During the follow-up, the patient developed conduct disorder of moderate severity, impulsivity and poor academic achievement with engagement in risk behaviours and substance use. His intellectual quotient (IQ) was 94 (normal-average). His brother exhibited adequate academic performance and achieved goals, albeit with considerable effort. Normal social skills and relationships. The brother refused the IQ evaluation. He required placement of a drain due to recurrent ear infections and surgery for recurrent kneecap luxation. The health records show normal serum cholesterol levels at ages 6-8 years (106-125 mg/dL). In a new evaluation, the results of array comparative genomic hybridization (aCGH) were normal; given that the patients were both of reproductive age and the suspicion of a similar syndrome with variable phenotypic expression without a specific suspected diagnosis, clinical genome sequencing was performed, leading to detection of the pathogenic variants c.452G>A and c.1A>G in the DHCR7 gene associated with SLOS.
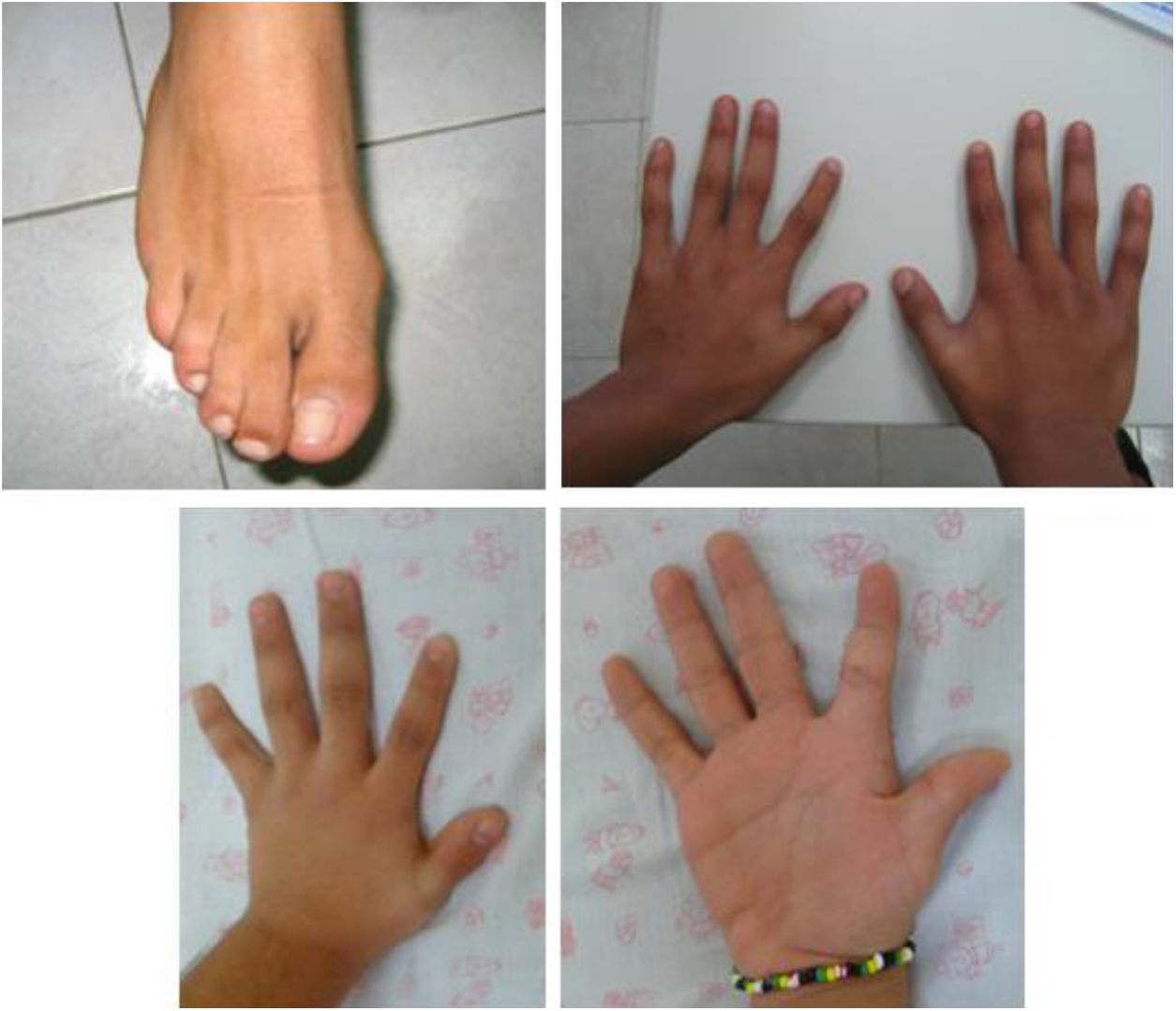
Table 1 summarises the clinical presentation and genotype of each case.
Summary of clinical characteristics and genotype of the cases.
| Case 1 | Case 2 | Case 3 | Case 4 | |
|---|---|---|---|---|
| Age at diagnosis (years) | 0 | – | 22 | 18 |
| Abnormalities in prenatal ultrasound | IUGR and oligohydramnios | Holoprosencephaly and abnormal sex development | IUGR | IUGR |
| Postnatal delayed growth | + | + | + | |
| Genitourinary malformations | Left-sided duplicated collecting system without dilatation | Ambiguous genitalia | – | – |
| Heart malformations | – | – | – | – |
| 2nd and 3rd toe syndactyly | + | + | + | |
| Cleft palate/bifid uvula | Cleft palate (soft palate) | Cleft palate | Bifid uvula | |
| Hearing loss | Moderate bilateral neurosensory | |||
| Developmental delay | Severe | Mild | – | |
| Intellectual disability | Severe | – | – | |
| Behavioural disorder | + | + | – | |
| Serum 7-dehydrocholesterol (μmol/L) | 436.3 | 52.32 | 10.16 | |
| Serum total cholesterol (mg/dL) | 27 | 145-170 | 106-125 | |
| DHCR7 gene sequencing | Pathogenic variants c.906C>G/c.964-1G>C | Pathogenic variants c.906C>G/c.964-1G>C | Pathogenic variants c.452G>A/c.1A>G | Pathogenic variants c.452G>A/c.1A>G |
The description of these cases illustrates the substantially heterogeneous expressivity of SLOS, even within families. The described biochemical profile supports the hypothesis that the syndrome is related to cholesterol transport from the mother to the embryo during pregnancy.5
In the prenatal period, SLOS should be suspected in the presence of de holoprosencephaly and ambiguous genitalia in XY foetuses, and in the postnatal period, it should be suspected in patients with intrauterine growth restriction, microcephaly, cleft palate and second-toe and third-toe syndactyly after ruling out chromosome disorders, even if the neurologic examination or the total cholesterol levels are normal.
Clinical exome sequencing is clearly useful for diagnosis of SLOS in the case of atypical phenotypes or classic but mild phenotypes, which has important repercussions not only for the patients, as it allows consideration of preventive and therapeutic interventions6 and adequate reproductive genetic counselling, but also for their families.
Please cite this article as: Sánchez-Soler MJ, Serrano-Antón AT, López-González V, Ballesta-Martínez MJ, Guillén-Navarro E. Expresividad extremadamente variable en el síndrome de Smith-Lemli-Opitz: revisión de 4 casos clínicos. An Pediatr (Barc). 2022. https://doi.org/10.1016/j.anpedi.2021.03.010
