



Langerhans cell histiocytosis (LCH) is characterised by the accumulation of dendritic cells in granulomatous lesions that mostly involve bone and skin.1 Gastrointestinal involvement is rare in LCH (1–5%), although its incidence is probably underestimated due to the nonspecific nature of its presenting symptoms and the association of the latter to the chronic course of disease or the treatments for it.2 We present two cases of LCH with gastrointestinal involvement.
Case 1Girl, 4 months of age presenting with a papular rash in the torso and scalp lasting 4 months associated with anorexia, delayed growth (3rd percentile) and haematochezia. The skin biopsy was consistent with LCH and displayed intense reactivity to CD1a and S100. After ruling out a cow's milk protein allergy, we performed a gastrointestinal endoscopy that showed nonspecific signs of duodenitis and colitis. The histological examination revealed a histiocytic infiltrate accompanied by eosinophils in the colorectal mucosa with immunohistochemical findings consistent with LCH (Fig. 1). The remaining staging studies ruled out disease at other locations. As a case of multisystem LCH without involvement of risk organs it was treated according to the LCH III group B protocol, consisting of a 6-week induction therapy with steroids and vinblastine, with improvement of the skin lesions and resolution of the colonic infiltrate. Subsequently, the patient completed a yearlong treatment course of steroids and vinblastine. The patient remained in remission 10 months after completing the treatment.
Case 2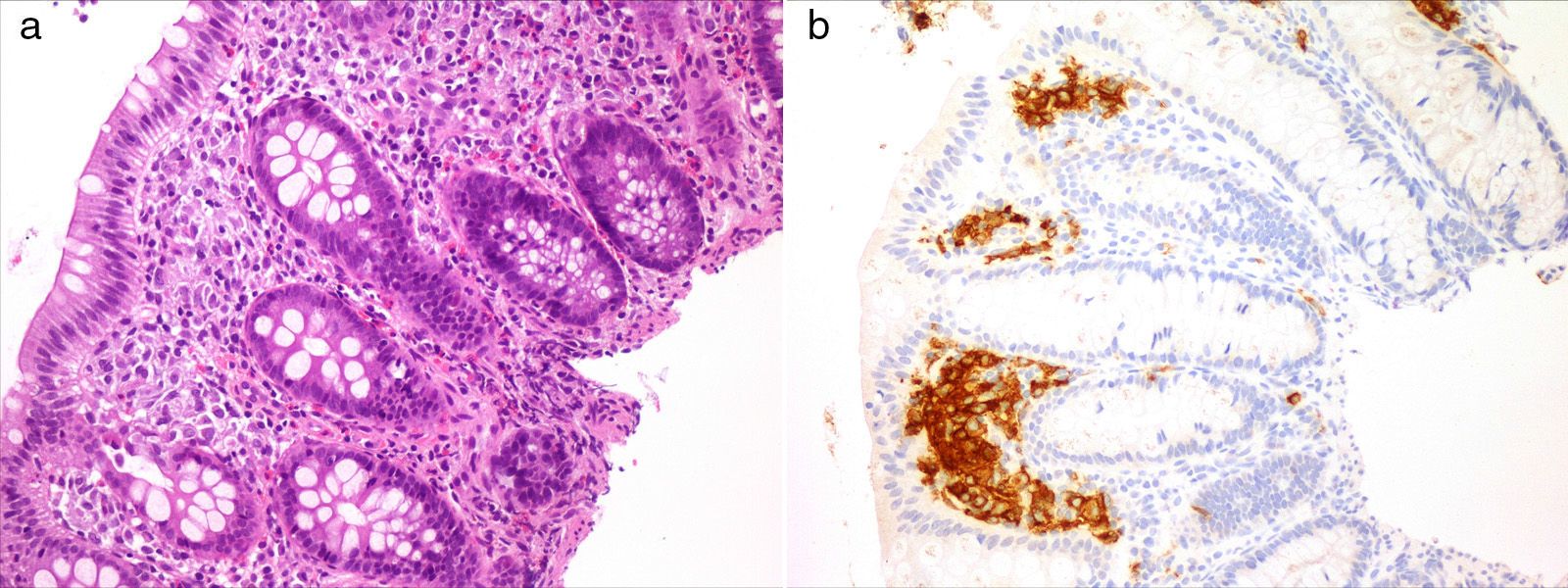
Girl, 12 years of age that presented with polyuria and polydipsia in the absence of fever and dysuria. The laboratory findings and water deprivation and desmopressin test were suggestive of central diabetes insipidus. Cranial magnetic resonance imaging showed a thickening of the pituitary stalk (78×10×11mm), with no changes in signal intensity. Blood and cerebrospinal fluid tested negative for the characteristic markers of germ cell tumours, leading to a working diagnosis of LCH, although a glial tumour could not be ruled out. Given the surgical risk involved in a biopsy, the case was managed with a watchful waiting approach. In subsequent months, the patient developed panhypopituitarism, with normal autoimmunity. The onset consisted of a prolonged fever with no constitutional symptoms and no abnormal findings in the physical examination. Testing for immune conditions, infectious diseases and cancer only yielded abnormal results for deaminated gliadin peptides, so an upper gastrointestinal endoscopy was performed to rule out celiac disease despite the absence of gastrointestinal symptoms. The histopathological examination revealed a histiocytic and eosinophillic infiltrate in the duodenal mucosa that tested positive for CD1a, S100 and langerin. After ruling out disease at other levels, the patient was diagnosed with LCH with pituitary and gastrointestinal involvement, and started chemotherapy as specified by the protocol for LCH III group B, with a favourable response. Two years after the end of treatment, the patient remained free of gastrointestinal symptoms and on replacement therapy for her panhypopituitarism.
Ninety-five percent of cases of LCH in the gastrointestinal tract are diagnosed in children less than 2 years of age, with a higher incidence in males. The typical presentation includes failure to thrive, diarrhoea, bloody stools, abdominal pain and vomiting.2 Patients with extensive involvement may develop malabsorption or protein-losing enteropathy (PLE), documented in up to 77% of cases. Bowel perforation has been reported less frequently.3,4 It most commonly involves the duodenum and colon in a multifocal pattern with superficial erosions or bleeding ulcers. Although cases with isolated gastrointestinal involvement have been described in the literature, gastrointestinal involvement usually occurs in the context of multisystem disease and is preceded by skin lesions in more than 80% of the patients.5 Cases of isolated gastrointestinal disease have been reported in adults, typically encountered as incidental solitary polyps in asymptomatic patients.6
The differential diagnosis basically includes dietary allergies, immunodeficiencies, inflammatory bowel disease and various causes of PLE. Although it is difficult to determine the prognosis of gastrointestinal involvement in children because it usually occurs with risk organ involvement, mortality can reach up to 55% at 18 months post diagnosis, especially in cases with associated PLE.2–4
Case 1 is a clear example of the typical presentation of the disease, with a history of delayed growth in an infant associated with skin lesions and gastrointestinal symptoms. The absence of associated PLE could be due to the favourable clinical outcome. Case 2 was exceptional in that it concerned an adolescent with no gastrointestinal symptoms or risk organ involvement. On the other hand, this case suggests that fever without a source may be another potential presenting symptom in LCH with gastrointestinal involvement.
Please cite this article as: Andión Catalán M, Ruano Domínguez D, Azorín Cuadrillero D, de Rojas de Pablos T, Madero López L. Histiocitosis de células de Langerhans con afectación gastrointestinal. An Pediatr (Barc). 2015;83:279–280.
