








Haemophagocytic syndrome, or haemophagocytic lymphohistiocytosis (HLH), is a disorder with high mortality, typically recognised at paediatric age. Without proper treatment, HLH can be fatal. The risk of a rapid progression to multi-organ failure and central nervous system involvement leading to long-term sequelae are the most feared consequences of a diagnostic delay. Therefore, HLH is a medical emergency that paediatricians should be able to identify in a patient with fever and progressive worsening of general condition. The application of the HLH diagnostic criteria include clinical and analytical data (as well as a bone marrow aspirate) and the search for a trigger (infectious, oncological, rheumatological, or metabolic). These are decisive for the establishment of a targeted treatment, which aims at neutralising the trigger and reducing the hyper-inflammation. The most relevant data for general paediatricians are presented in this review, including the physiopathology, diagnosis, and treatment of this serious disease.
El síndrome hemofagocítico (SHF) o linfohistiocitosis hemofagocítica es una entidad con elevada mortalidad, típicamente reconocida en la edad pediátrica. Sin un correcto tratamiento, el SHF puede ser fatal: el riesgo de una rápida progresión a fallo multiorgánico y de afectación del sistema nervioso central con secuelas a largo plazo, son las consecuencias más graves de un retraso diagnóstico. Por lo tanto, el SHF es una urgencia médica que los pediatras deben saber identificar en un paciente con fiebre y afectación progresiva del estado general. La aplicación de los criterios diagnósticos de SHF, que consideran datos clínicos y analíticos (incluyendo un aspirado de médula ósea), y la búsqueda del factor desencadenante (infeccioso, oncológico, reumatológico, metabólico), son claves para poder instaurar un tratamiento dirigido, que neutralice el desencadenante y frene la hiperinflamación. En la presente revisión se exponen los datos más relevantes sobre la fisiopatología, diagnóstico y tratamiento de esta grave enfermedad para pediatras generales.
Haemophagocytic lymphohistiocytosis (HLH), also known as haemophagocytic syndrome (HPS), is a syndrome caused by an abnormal immune response to a trigger that may be of an infectious, oncologic, rheumatologic, or metabolic nature, giving rise to a state of hyperinflammation. This hyperinflammatory response causes a massive release or storm of cytokines, which in turn is responsible for the clinical manifestations. HLH is not a single disease, but a syndrome that may be caused by any of a wide range of underlying conditions that cause a specific and characteristic inflammatory phenotype.1,2
The term haemophagocytosis refers to the characteristic findings associated with macrophage activation, with engulfment of red blood cells, white blood cells, platelets, and precursors. This phenomenon may occur in other clinical situations and results in the destruction (digestion) of blood cells in the bone marrow and other tissues. When haemophagocytosis occurs in the context of a highly stimulated but inefficient immune response, it is known as HLH. This hyperimmune response is associated with infiltration of activated macrophages and lymphocytes in multiple organs, and examination of biopsy specimens of affected tissues reveals signs of inflammatory events and haemophagocytosis.
HLH may develop at any age, although the primary form of disease usually has onset in childhood. It was initially described in association with viral infection, but it has been subsequently found in association with fungal, bacterial, and parasitic infections (specifically leishmaniasis).2,3 Infection, especially by Epstein–Barr virus (EBV), is a common trigger for both primary and secondary HLH. It may also occur in the context of autoimmune disorders such as juvenile idiopathic arthritis, systemic lupus erythematosus, or Kawasaki disease.4 In fact, any strong activation of cellular immunity (due to infection, rheumatism, malignant disease, etc.) may trigger a secondary form of HLH. In the context of rheumatic disease, HLH is known as macrophage activation syndrome.
Both primary and secondary HLH may have a rapidly fatal course and require early treatment.5–7
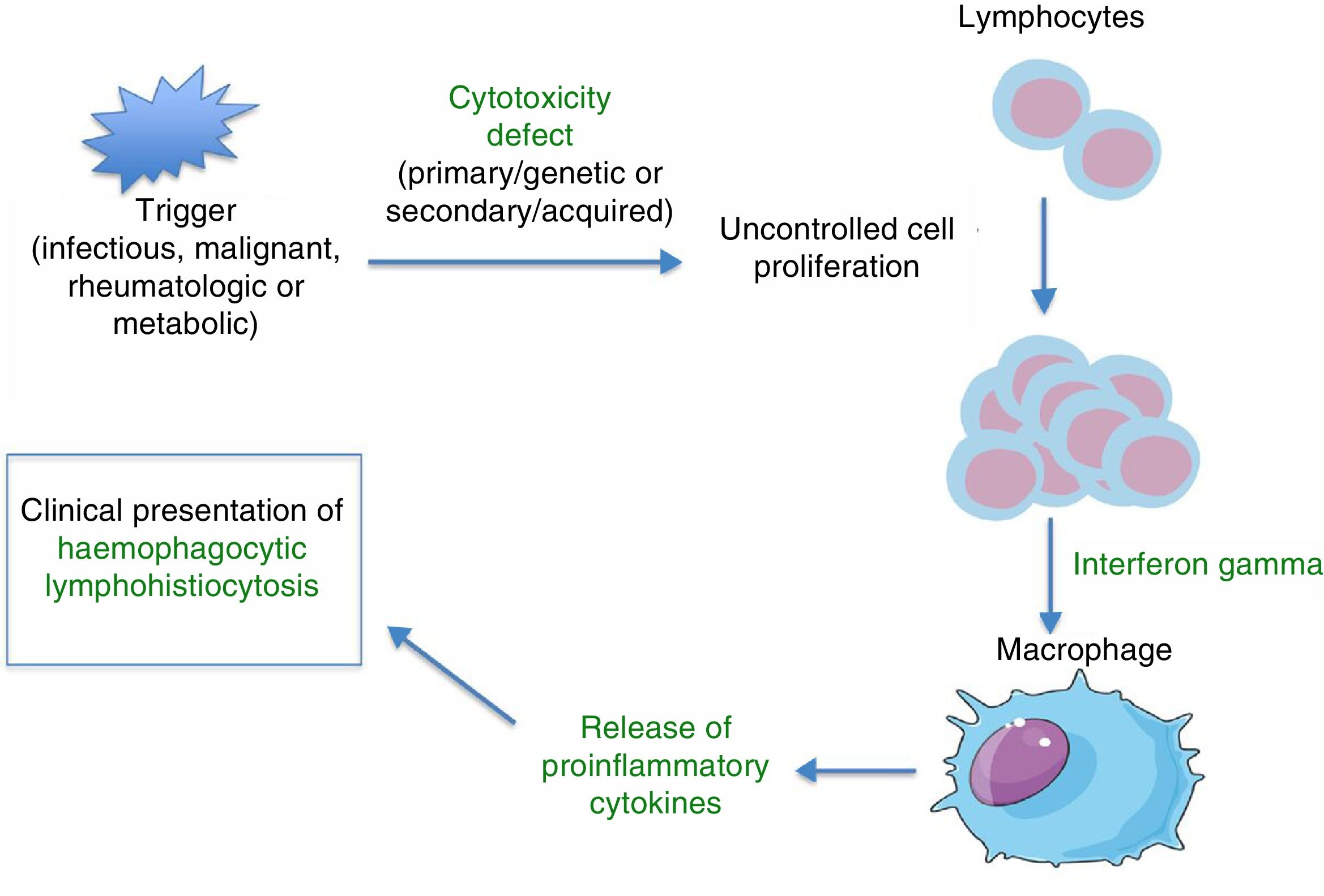
PathophysiologyHLH is a severe hyperinflammatory state caused by the proliferation and activation of lymphocytes and macrophages, which release massive amounts of cytokines. This process is frequently triggered by an infectious agent, which activates an immune system that is dysregulated either due to a primary/genetic defect (primary HLH) or secondary to an underlying disease (autoimmune, autoinflammatory, malignant, or metabolic) or immunomodulatory treatments. Several studies on murine models8 have demonstrated that abnormalities in the cytotoxic function of CD8+ lymphocytes play a key role in the pathophysiology of HLH. The physiological function of the cytotoxic response mediated by CD8+ and NK cells is the lysis of target cells (infected, malignant, aged, or overabundant) through the exocytosis of cytotoxic granules (known as degranulation), which contain cytolytic proteins (granzymes) and perforins (proteins that create a pore in the plasma membrane of the target cell through which the granzymes can enter the cell to lyse it). Deficient removal of target cells stimulates antigen presentation by dendritic cells and perpetuates T-lymphocyte activation. The sustained and uncontrolled activity of these lymphocytes, which release large amounts of interferon gamma (IFN-γ), causes macrophage activation, which in turn leads to haemophagocytosis, with release of proinflammatory cytokines (TNF-α, IFN-α, IL-6, IL-18, IL-12) that again perpetuate antigen presentation and T-cell activation, all of which leads to the clinical manifestations of HLH (Fig. 1 and Table 1). This produces a vicious cycle of inflammation and cytokine release. Since HLH involves defects in cytotoxicity, cytotoxic function is usually assessed for diagnosis and generally found to be low to absent in both primary and secondary forms. The difference is that in primary forms, the cytotoxicity defect persists even in the absence of HLH symptoms. There is evidence of a correlation between the severity of disease and residual cytotoxic function, so that the intensity of symptoms may vary based on the genetic defect, type of mutation, and triggering agent. Lastly, the pathophysiological mechanisms of secondary forms of HLH are not clearly understood. We know that not every infectious agent can trigger HLH—infectious agents require specific characteristics to have this effect. Chief among the causative agents is EBV, which can interfere with the activity of CD8+ cells through specific proteins, leading to the massive release of cytokines, mainly IL-18 and IFN-γ.1,2
Clinical and laboratory features. Underlying mechanisms.
| Cardinal manifestations | |
|---|---|
| Manifestations | Underlying mechanism |
| Fever | Elevation of IL-1 and IL-6 |
| Pancytopenia | Elevation of TNFα and haemophagocytosis |
| Hypertriglyceridemia | TNFα inhibits lipoprotein lipase |
| Hyperferritininaemia | Ferritin is secreted by macrophages activated by IFN-γ |
| Hypofibrinogenemia | Plasminogen activators are secreted by activated macrophages and cause hyperfibrinolysis |
| High levels of sCD25 (soluble interleukin-2 receptor) | Activated lymphocytes are the source of sCD25 |
| Hepatosplenomegaly | Infiltration of multiple organs by activated lymphocytes and macrophages (histiocytes) |
| Other manifestations |
|---|
| Clinical: lymphadenopathies, liver failure, bleeding diathesis, oedema, neurologic manifestations, multiple organ failure |
| Laboratory: leukopenia (lymphopaenia), hypoproteinaemia, hypoalbuminemia, hyponatraemia, hypertransaminasaemia, hyperbilirubinaemia, LDH elevation, D-dimer elevation |
Adapted from Brisse et al.8
At onset, the clinical manifestations of HLH are those of any severe infectious disease. It is characterised by prolonged high fever and progressive pancytopaenia and hepatosplenomegaly, with other signs of multiple organ failure with potential involvement of the liver, lungs, central nervous system (CNS), etc.6,7 Some patients present with hair loss and hypoalbuminaemia. The laboratory findings include cytopaenias, coagulopathy, hypertriglyceridemia, hypofibrinogenemia, hypertransaminasaemia, and hyperferritinaemia7 (Table 1). These findings constitute the clinical and laboratory criteria used for diagnosis, first established in 1991 and reviewed in 20042,4,6 (Table 2). Bone marrow aspirate specimens are usually hypercellular with normal cell maturation.
Clinical criteria for diagnosis of haemophagocytic lymphohistiocytosis (HLH-2004 guidelines).
| Familial HLH or HLH with a known genetic cause |
| Clinical and laboratory criteria (at least 5 out of 8 must be met for diagnosis) |
| 1. Fever ≥38.5°C |
| 2. Splenomegaly |
| 3. Cytopaenias |
| Haemoglobin <90g/L (if age < 4 weeks, <120g/L). |
| Platelets < 100 000/mm3 |
| Neutrophils <1000/mm3 |
| 4. Hypertriglyceridemia and/or hypofibrinogenemia |
| Fasting triglycerides ≥3mmol/L |
| Fibrinogen <1.5g/L |
| 5. Ferritin >500μg/l |
| 6. sCD25 ≥ 2400U/ml |
| 7. Low or absent NK-cell activity |
| 8. Haemophagocytosis in bone marrow, CSF, or lymph nodes |
Source: Henter et al.6
HLH may present with neurologic symptoms due to CNS infiltration by activated macrophages. The presentation in these cases resembles that of encephalitis. Analysis of the cerebrospinal fluid (CSF) reveals mononuclear cell pleocytosis (lymphocytic meningitis) or albuminocytologic dissociation. In cases with CNS involvement, HLH has a significantly worse prognosis and may cause permanent neurologic sequelae.
Not all diagnostic criteria are present at onset or in newborns. Haemophagocytosis is not required for diagnosis. It is the progression of the criteria that should alert physicians of the possible presence of HLH.1 Diagnosis is challenging, as it is difficult to distinguish between the physiological macrophage activation that occurs in sepsis,9 malignancies, or autoimmune/autoinflammatory diseases and the pathological activation that characterises HLH.10
In short, a poor response or unusual progression of symptoms of common diseases should lead to suspicion of HLH. Although microbiological findings may confirm the presence of infection, the latter may be a trigger of HLH, which would lead to progressive multiple organ failure, would not respond to customary antimicrobial treatment and would require specific treatment. Therefore, early diagnosis and treatment are associated with better outcomes.
Diagnosis and differential diagnosisAccording to the clinical and laboratory criteria proposed by the Histiocyte Society6 (Table 2), HLH can be diagnosed when patients meet five or more of the eight clinical criteria or when there is a confirmed molecular diagnosis. An evaluation of immune function and genetic testing are important, especially in the diagnosis of familial or primary forms.
Broadly speaking, fever and splenomegaly are found in 90–100% of affected children, excepting newborns.1,4 Hyperferritinaemia is a significant marker, especially when ferritin levels are very elevated (>10 000μg/L in children, with a sensitivity of 90% and a specificity of 96%).4,7 Haemophagocytosis is detected in bone marrow in only 30–40% of cases at onset, which hinders diagnosis, and its presence increases to 80–90% of patients as the disease progresses.1 The serum level of soluble interleukin-2 receptor (sIL-2r or sCD25) is one of the diagnostic criteria (level>2400U/mL), since it is a marker of lymphocyte activation. Recent studies have shown that the ratio of the serum levels of sCD25 and ferritin may be useful to differentiate specific forms of HLH.11 Assessment of NK activity is the technique that applies to the widest range of disease forms, as changes in cytotoxic activity are characteristic of HLH in general. Unfortunately, there are barriers to ordering the assessment of NK activity and determination of serum levels of sCD25, as these tests are only available in specialised immunology laboratories.
One of the greatest challenges posed by HLH is its diagnosis, as its initial signs and symptoms are non-specific (Fig. 2). On account of its clinical picture, it should be included in the differential diagnosis of fever of unknown origin, acute liver failure, or hepatitis with coagulopathy (30% of patients present with elevated transaminase levels >100U/L),12 sepsis with multiple organ failure, lymphocytic meningitis or encephalitis with focal CNS lesions, and, in the neonatal period, isolated CNS involvement or fulminant liver failure mimicking metabolic disease.
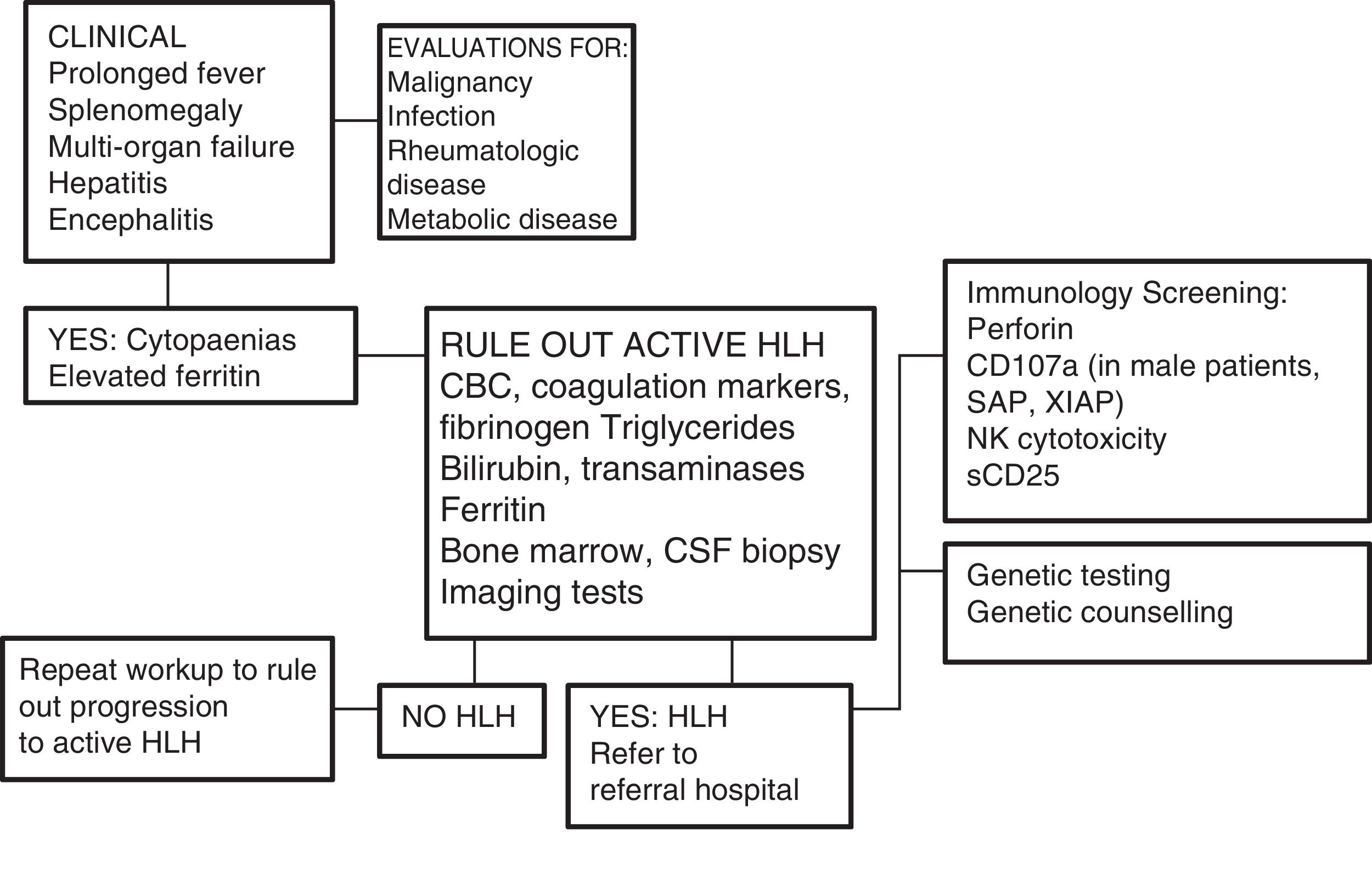
Diagnostic algorithm. Adapted from the Cincinnati Children's HLH Diagnostic Strategy, available at: www.cincinnatichildrens.org/dchi.
Analysis of the proteins involved in the pathophysiology of HLH by flow cytometry can be very useful, for instance, in cases of perforin deficiency (FLH-2), SAP deficiency (XLP-1), or XIAP deficiency (XLP-2) (Fig. 2), although these criteria are not yet included in the HLH-2004 protocol.6
In recent years, research has identified several genetic mutations that affect lymphocyte cytotoxicity and that are the main causes of primary HLH.1,4,13,14 But there are still cases of suspected primary HLH (up to 30%), even with a family history or consanguinity, in which a causal defect has not been identified,15 and it is believed that there are other genes involved that have yet to be found. The recent literature includes descriptions of cases with heterozygous or combined FHL mutations.14
Primary forms include familial haemophagocytic lymphohistiocytosis (FHL), immunodeficiencies associated with oculocutaneous albinism, and lymphoproliferative syndromes associated with EBV infection (Table 3). Mutations have been found in 12 different genes, with autosomal recessive or X-linked patterns of inheritance. Patients with HLH are genetically heterogeneous, but tend to share a similar immunophenotype due to the defect in cytotoxicity.13,14
Genetic forms of HLH.
| Gene | Protein | Disease |
|---|---|---|
| Familial HLH | ||
| PRF1 | Perforin | Perforin deficiency, or FHL2 |
| UNC13D | Munc13-4 | Munc13-4 deficiency, or FHL3 |
| STX11 | Syntaxin 11 | Syntaxin 11 deficiency, or FHL4 |
| STXBP2 | Munc18-2 | Munc18-2 deficiency, or FHL5 |
| Immunodeficiencies associated with hypopigmentation | ||
| RAB27A | Rab27a | Griscelli syndrome type 2 |
| LYST | Lyst | Chediak-Higashi syndrome |
| AP3B1 | AP3B1 | Hermansky-Pudlak syndrome type 2 |
| EBV-associated lymphoproliferative syndromes | ||
| SH2D1A | SAP | SAP deficiency, or XLP1 |
| XIAP | XIAP | XIAP deficiency, or XLP2 |
| ITK | ITK | ITK deficiency |
| CD27 | CD27 | CD27 deficiency |
| MAGT1 | Magnesium transporter protein 1 | MAGT1 deficiency or XMEN disease |
Mutations in the PRF1 gene (FHL2) encoding the perforin protein were the first to be identified in relation with HLH. Perforin deficiency amounts to 35%15 of molecular abnormalities, although the proportion varies between populations. Flow cytometric perforin screening is highly sensitive and specific.
Mutations in the UNC13D gene cause Munc13-4 deficiency, known as FHL3 (35% of cases of FHL). In southern Europe, FHL2 and FHL3 combined amount to nearly 70% of cases.15 Syntaxin 11 deficiency (FHL4) accounts for only 5% of the total cases of FHL, and last of all, Munc18-2 deficiency (FHL5) accounts for only 5–20% of the total. The FHL3, 4, and 5 forms present with severe impairment of NK cytotoxicity, which makes the evaluation of NK cell function essential.12,16
Immunodeficiencies associated with oculocutaneous albinismGriscelli syndrome type 2, Chediak–Higashi syndrome or Hermansky–Pudlak syndrome type 2 (HP2) manifest with defects in the traffic and exocytosis of secretory lysosomes (which play important roles in numerous cell populations), leading to neurodegenerative diseases, hypopigmentation or albinism, coagulopathy, neutropenia, and abnormalities in NK activity. These individuals usually have silver or whitish-greyish hair whose analysis with a light microscope may guide the diagnosis. The presence of vacuolated neutrophils in peripheral blood is suggestive of Chediak–Higashi syndrome. Palmoplantar keratoderma is suggestive of HP2. This type of primary immunodeficiencies may manifest as HLH.17
Lymphoproliferative syndromes associated with Epstein–Barr virusThere is evidence of a strong association in male patients between EBV-associated HLH and X-linked lymphoproliferative syndromes (XLP1 and XLP2, whose markers, the proteins SAP [XLP1] or XIAP [XLP2] can be assessed by flow cytometry, and MAGT1 deficiency).18 The literature also describes defects in ITK and CD27 with an autosomal recessive pattern of inheritance. Some cases may be initially diagnosed as infectious mononucleosis due to EBV, but they progress and eventually turn into severe HLH.3
Acquired or secondary HLH (Table 4)Secondary HLH is clinically identical to primary HLH, but there is no family history of HLH or a genetic defect associated with impaired cytotoxicity. Numerous cases have been described, mainly in association of infections of any type (viral, bacterial, fungal, or parasitic), autoinflammatory or autoimmune diseases, malignancies, and metabolic disorders. Virtually any uncontrolled infection may lead to a clinical picture compatible with HLH. We must specifically mention infection by Leishmania parasites, which should always be ruled out by testing of a peripheral blood or bone marrow specimen (staining or PCR).
Most frequent causes of secondary or acquired HLH.
| Infections | Malignancy | Rheumatologic disease | Diseases |
|---|---|---|---|
| Viruses (EBV > HSV, CMV, hepatotropic viruses, dengue, enterovirus, PVB19) | Lymphoma | Juvenile idiopathic arthritis | Lysinuric protein intolerance |
| Bacteria and mycobacteria | Leukaemia | Systemic lupus erythematosus | Biotinidase deficiency |
| Fungi | Melanoma | Vasculitis (PAN) | Mevalonic aciduria |
| Parasites (Leishmania) | Other tumours | Autoinflammatory diseases (NLRC4) Kawasaki disease | Transaldolase deficiency Gaucher disease Wolman disease |
In other cases, HLH is secondary to other immunodeficiencies caused by chemotherapy, immunosuppressive drugs, biologic drugs, and transplantation.1,13 It can develop at any age in association with malignant disease, at the time of onset and diagnosis or during its treatment.18
Rheumatological diseases such as juvenile idiopathic arthritis, systemic lupus erythematosus, or NLRC4 deficiency frequently cause secondary HLH, mainly at onset or due to poor disease control. In this context, HLH is usually referred to as macrophage activation syndrome.9
Broadly speaking, in acquired HLH the age of onset tends to be older compared to primary HLH, the trigger is identified, and recurrence is less likely.19
Immunology testing (Fig. 2)The main immunology laboratory tests for diagnosis of HLH are based on flow cytometry techniques:
- 1.
Intracellular perforin stain (screening for FHL2).
- 2.
CD107a degranulation assay (screening for FHL3-5 and cases without a molecular diagnosis).
- 3.
NK cell function testing (assesses overall impairment).
Of these tests, the combination of techniques 1 and 2 is superior to NK cell function testing (lower specificity and sensitivity).20 Immunology tests can differentiate between primary and secondary forms,13 as they have different patterns of T-cell activation, differentiation, and degranulation.13,21 However, these techniques do not offer immediate results, so initial treatment decisions must be made based on the severity of symptoms.6
TreatmentThe general goal of treatment is the suppression and control of hyperinflammation and elevated cytokine levels and the elimination of activated and infected cells. In genetic forms, the only curative treatment that can correct the cytotoxicity defect is haematopoietic stem-cell transplantation (HSCT). The possible treatment modalities include corticosteroid drugs (the first step of treatment per the HLH-94 and HLH-2004 protocols, usually dexamethasone at 10mg/m2 in primary forms and methylprednisolone in secondary forms), immunosuppressive drugs, cytostatic drugs, immunomodulators, monoclonal antibodies, and anticytokine therapy (Table 5).5,6
Treatment should be based on the severity and course of disease. There are very severe forms with a fulminant course that require specific treatment and intensive supportive care on an empirical and urgent basis, while milder or recurrent cases may respond to less aggressive treatment.3,5
Therapeutic decision-making is complex, as many patients present with infections with an identified aetiological agent and antimicrobial treatment is prioritised that fails to halt the inflammatory cascade. Although it may seem paradoxical, these patients need to receive immunosuppressive agents such as steroids, cytokine blockers, immunoglobulin therapy, or even cytostatic drugs to destroy the activated cells. Severe forms require admission to paediatric intensive care units, as the patients need mechanical ventilation, haemodynamic support, transfusions, etc. In such severe cases, consultation with specialists and transfer of the patient to a tertiary care hospital is recommended.5
An exhaustive microbiological investigation for identification of the causative agent and administration of appropriate antibacterial, antifungal, antiviral, or antiparasitic treatment are of the essence.1,3,5,6 In most cases, appropriate anti-infective therapy is not sufficient and must be combined with specific treatment for HLH, except in cases of leishmaniasis, which usually resolve with antimicrobial treatment, such as liposomal amphotericin B.
In severe and complex cases, initiation of treatment based on clinical suspicion is recommended even if the patient does not strictly meet the necessary diagnostic criteria, as HLH is an urgent and life-threatening condition. Moderate forms tend to respond well to corticosteroid and immunoglobulin therapy, but the most severe forms require a combination of immunosuppressive or cytotoxic drugs.
Without treatment, the prognosis of HLH is bleak, especially in genetic forms.22 The introduction of etoposide in the 1990s was a significant advance in treatment. We ought to mention the results of the protocol of the HLH-94 collaborative study23 (Histiocyte Society), which was based on a combination of dexamethasone, etoposide, and cyclosporin A followed by HSCT in patients with familial, recurrent, or severe and persistent forms. This protocol succeeded in improving overall survival, with an estimated 5-year probability of survival of 54%, although 29% of patients died before undergoing HSCT. The favourable results were sustained in the long-term,24 although 22% of patients developed neurologic sequelae.25
The following study, the HLH-2004,7 demonstrated the benefits of steroid therapy and etoposide in a cohort of 369 children from 27 countries, including Spain.26 The proposed changes—administration of ciclosporin A from the beginning of treatment and recommendation of HSCT when a suitable donor was found—did not achieve significant improvements in survival (5-year survival, 62%) or decreases in early mortality (19%). The 5-year post-HSCT survival was similar (67% in HLH-2004, 66% in HLH-94).23,26 The authors identified predictors of poor outcome: neurologic involvement and pleocytosis in CSF, and elevated levels of bilirubin and ferritin at diagnosis.27 No collaborative studies have been proposed since 2012, and at present we recommend using the HLH-94 protocol with the HLH-2004 diagnostic criteria.
Other studies have achieved good outcomes with antithymocyte globulin (ATG),28 alemtuzumab (anti-CD52 antibody),29 or rituximab (anti-CD20 antibody).30 Clinical trials of anti-interferon gamma monoclonal antibodies and hybrid immunotherapy (ATG, dexamethasone, and etoposide) are underway, and publication of results is still pending. There are reports of a favourable response of refractory forms to salvage therapy with anakinra, alemtuzumab, tocilizumab, etanercept, or ruxolitinib.29
The duration of treatment depends on the progress and response of the patient. In primary forms, the initial treatment prepares the ground for transplantation: its aim is to achieve remission while searching for the most suitable donor. In secondary forms, treatment may last a few days or weeks if clinical remission is achieved, followed by monitoring for potential recurrences. If HLH recurs, targeted treatment may need to be reinitiated and transplantation considered.
In familial or primary forms, allogeneic HSCT is considered the only curative treatment available to reconstitute the impaired immune system. It is also recommended in very severe, progressive, or recurrent cases, and even in asymptomatic siblings with a confirmed molecular diagnosis. The complications associated with HSCT tend to be considerable, so it is recommended that this procedure be performed in centres with experience in the technique and using reduced intensity conditioning regimens.25,31 Stable mixed donor chimerism appears to be sufficient to prevent recurence.31
Recommendations for secondary HLH are still vague, although prolonged treatment should be avoided. Severe forms with an infectious trigger, such as EBV, may require dose-intense therapy with etoposide or even HSCT.3,5 The management of HLH associated with malignancies is very complex due to the need to fight cancer and HLH at the same time.32 In macrophage activation syndromes, 33 the proposed first line of treatment is high-dose steroid therapy with cyclosporin, with anakinra (an interleukin-1 inhibitor) as the second-line drug and exceptional use of etoposide.
ConclusionsHLH is associated with a high mortality and usually identified during childhood.
Without adequate treatment, HLH may be fatal. Rapid progression to multiple organ failure and CNS involvement with long-term sequelae are the direst consequences of diagnostic delay.
Consequently, HLH is a medical emergency that paediatricians must be able to identify in patients presenting with fever and progressive deterioration of general health.
The application of the HLH diagnostic criteria, which include clinical and laboratory parameters, and the investigation of the trigger (infectious, malignant, rheumatologic, metabolic) are key, as they allow delivery of targeted treatment with the purpose of neutralising the trigger and halting the hyperinflammatory process.
FundingThis work was funded in part by grants given by the Fondo de Investigaciones Sanitarias (FIS) to Luis M. Allende and Luis I. González-Granado (PI 16/2053) and to Juana Gil Herrera and Itziar Astigarraga (PI12/02761), as well as funds received from the televised rare diseases fundraiser marathon of the EITB/BIOEF for the Histocytosis project of Astigarraga (BIO16/ER/020/BC). It was also supported by funds from the ERDF.
Conflicts of interestThe authors have no conflicts of interest to declare.
We thank the patients, their families, and the professionals that have participated in the collaborative investigations. We also thank Susana García-Obregón for her contribution on behalf of Spain to the international histiocytosis cooperative study.
Please cite this article as: Astigarraga I, Gonzalez-Granado LI, Allende LM, Alsina L. Síndromes hemofagocíticos: la importancia del diagnóstico y tratamiento precoces. Anales de Pediatría. 2018;89:124.e1–124.e8.
