



Treatments with CFTR protein modulators have improved respiratory and digestive health in patients with cystic fibrosis.
ObjectiveTo assess changes in intestinal inflammation through the analysis of fecal calprotectin in patients with cystic fibrosis during treatment with CFTR modulators.
Material and methodsProspective multicenter study of changes in fecal calprotectin in patients with cystic fibrosis treated with CFTR modulators, comparing double combinations (lumacaftor/ivacaftor or tezacaftor/ivacaftor) and triple combinations (elexacaftor/tezacaftor/ivacaftor). We collected aata before treatment initiation and at 6 and 12 months.
ResultsAnalysis of 117 patients (69% with F508del/F508del). The median baseline fecal calprotectin level was 49 µg/g (IQR, 23–108); 48.7% had median levels greater than 50 µg/g and 11% levels greater than 250 µg/g. Fecal calprotectin decreased in both groups, with a greater decrease in patients treated with elexacaftor/tezacaftor/ivacaftor. We found a progressive decrease in abnormal values (>50 µg/g) at 6 months (48.7% vs 33.1%; P = .0067) and at 12 months (54% vs 33.5%; P = .0218). In the elexacaftor/tezacaftor/ivacaftor group, only two patients at 6 months and one patient at 12 months had levels greater than 250 µg/g. The estimated change at 12 months in the triple therapy group compared to the other group was −133 µg/g (95% CI, −254 to −13; P = .030); and, adjusting for sex, probiotics and Pseudomonas aeruginosa, −130 µg/g (−259 to −1; P = .049).
ConclusionsTreatment with CFTR modulators reduces intestinal inflammation in patients with cystic fibrosis, with a greater decrease in patients treated with triple therapy.
Los tratamientos con moduladores de la proteína CFTR han mejorado la salud respiratoria y digestiva de los pacientes con fibrosis quística.
ObjetivoAnalizar los cambios en la inflamación intestinal mediante estudio de la calprotectina fecal en pacientes con fibrosis quística durante el tratamiento con moduladores.
Material y métodosEstudio multicéntrico prospectivo de los cambios en la calprotectina fecal de pacientes con fibrosis quística tratados con moduladores, comparando aquellos constituidos por 2 moléculas (lumacaftor/ivacaftor o tezacaftor/ivacaftor), con los de 3 moléculas (elexacaftor/tezacaftor/ivacaftor). Se recogen datos antes de iniciar el tratamiento, a los 6 y a los 12 meses.
ResultadosAnálisis de 117 pacientes (69% F508del/F508del). Mediana basal de calprotectina fecal 49 mcg/g (RIC 23–108); 48.7% con >50 mcg/g; 11% con >250 mcg/g. Hubo disminución de calprotectina fecal en ambos grupos, siendo mayor en aquellos con elexacaftor/tezacaftor/ivacaftor. Se observó una disminución progresiva de los valores anormales (>50 mcg/g) a los 6 meses 48.7% vs 33,1% (p = 0,0067) y a los 12 meses 54% vs 33,5% (p = 0,0218). En el grupo elexacaftor/tezacaftor/ivacaftor, sólo 2 pacientes a los 6 meses y 1 paciente a los 12 meses, tenían >250 mcg/g. El cambio estimado (IC del 95%) a 12 meses en el grupo con triple terapia en comparación con el otro grupo fue de −133 mcg/g (−254−13, p = 0,030); y ajustado por sexo, probióticos y Pseudomonas aeruginosa de −130 mcg/g (−259 to −1, p = 0,049).
ConclusionesEl tratamiento con moduladores CFTR reduce la inflamación intestinal en pacientes con fibrosis quística, siendo la mejoría más destacada en pacientes tratados con la triple terapia.

Cystic fibrosis (CF) is a disease caused by changes in the gene that encodes the cystic fibrosis transmembrane conductance regulator protein (CFTR),1 found in the membrane of exocrine glands. Out of the more than 2000 CFTR variants known to date, the most frequent one is F508del, corresponding to the loss of an amino acid, phenylalanine, at position 508. The CFTR protein is responsible for chloride transport in the cell membrane, and changes in it result in changes in ion concentrations leading to dehydration of secretions in affected organs. The associated increase in morbidity and mortality is associated with the development of chronic obstructive pulmonary disease, which determines the prognosis and survival of individuals with CF.2
The recent introduction of CFTR modulators has been a considerable advance in the treatment of CF, achieving a substantial improvement in function in the abnormal protein.3–5 Ivacaftor (IVA), lumacaftor (LUM), tezacaftor (TEZ) and elexacaftor (ELE) are the molecules responsible for these striking changes.6 They are available as combinations in a single tablet as LUM/IVA, TEZ/IVA (dual combinations) or ELE/TEZ/IVA (ETI) (triple combination).6 The efficacy of the earlier combinations developed to treat patients homozygous for F508del (LUM/IVA o TEZ/IVA) was promising, but the outcomes were modest in clinical trials and real-world practice. However, the ETI combination, referred to as “triple therapy”, for treatment of patients carrying the F508del variant in at least one allele, has exhibited a significantly greater efficacy than its predecessors.7 Treatment with CFTR modulators decreases the frequency of respiratory exacerbations and improves lung function and quality of life in affected patients, drastically changing the course and prognosis of the disease.8–10 There is still a dearth of evidence on the changes in nutritional, anthropometric11 or gastrointestinal outcomes associated with the use of CFTR modulators, but a growing number of studies have found improvements in other aspects, such as a reduction in intestinal inflammation.
Fecal calprotectin (FC) is a biomarker of intestinal inflammation and its levels tend to be higher in patients with CF compared to healthy controls. This elevation may be due to chronic bowel inflammation resulting from the accumulation of thickened mucus, changes in the intestinal microbiota due to the use of antibiotics or to recurrent respiratory infections, among other possible causes. Multiple factors contribute to the elevation of FC in the context of CF, reflecting the complexity of this disease, which affects both the respiratory and the digestive systems.12,13
We conducted a prospective study in 15 pediatric cystic fibrosis reference units in Spain to assess changes in intestinal inflammation through the measurement of FC in patients with cystic fibrosis treated with LUM/IVA, TEZ/IVA and ETI at three time points: before treatment and at 6 and 12 months of treatment.
Material and methodsStudy designWe conducted a multicenter prospective cohort study in patients with CF treated with CFTR modulators, classifying participants in two groups: patients treated with LUM/IVA or TEZ/IVA (dual combinations) and patients treated with ETI. Fifteen pediatric CF units in Spanish hospitals participated in the study. The study was approved by the Ethics Committee of the hospital to which the principal investigator was affiliated (ref. 282−19), a decision ratified by the committees of the remaining participating hospitals. All participants aged 12–18 years provided their assent to participation, and adult patients and the parents of minors provided signed informed consent to participation. The inclusion criteria were having a diagnosis of CF and initiating treatment with CFTR modulators. All study visits took place in outpatient settings and in the context of routine checkups.
Clinical and demographic characteristicsWe collected data on demographic and clinical variables: sex, age at treatment initiation, pancreatic sufficiency, genotype, meconium ileus and presence of liver disease associated with cystic fibrosis or abnormal glucose levels. We analyzed FC levels before initiation of CFTR modulator therapy and at 6 and 12 months of treatment; we did not take into account the technique used in each center, as we assessed changes relative to baseline in each of the patients.
Statistical analysisWe collected and managed data using the Research Electronic Data Capture (REDCap) platform (11), data management digital tools hosted by the Sociedad Española de Gastroenterología, Hepatología y Nutrición Pediátrica (SEGHNP, Spanish Society of Gastroenterology, Hepatology and Nutrition). We received technical support from the AEGREDCap Support Unit, shared with the Asociación Española de Gastroenterología (AEG, Spanish Association of Gastroenterology). REDCap is a validated web-based software system designed to support data capture for research that provides an intuitive interface for validated data collection, auditing systems to track data handling and automated export and import features to download data to commonly used statistical software packages and upload data from external sources. The statistical analysis was performed with the Stata software package, version 13.0. The analysis was stratified by group based on the type of modulator therapy: LUM/IVA or TEZ/IVA group and ETI group. We expressed demographic, clinical and FC values as median and interquartile range (IQR), since the data were not normally distributed (determined by means of the Kolmogorov-Smirnov test) or as percentages. We assessed differences in FC levels after treatment initiation using the Wilcoxon signed rank test or the McNemar test for paired proportions. In every test, we considered p values of less than 0.05 statistically significant.
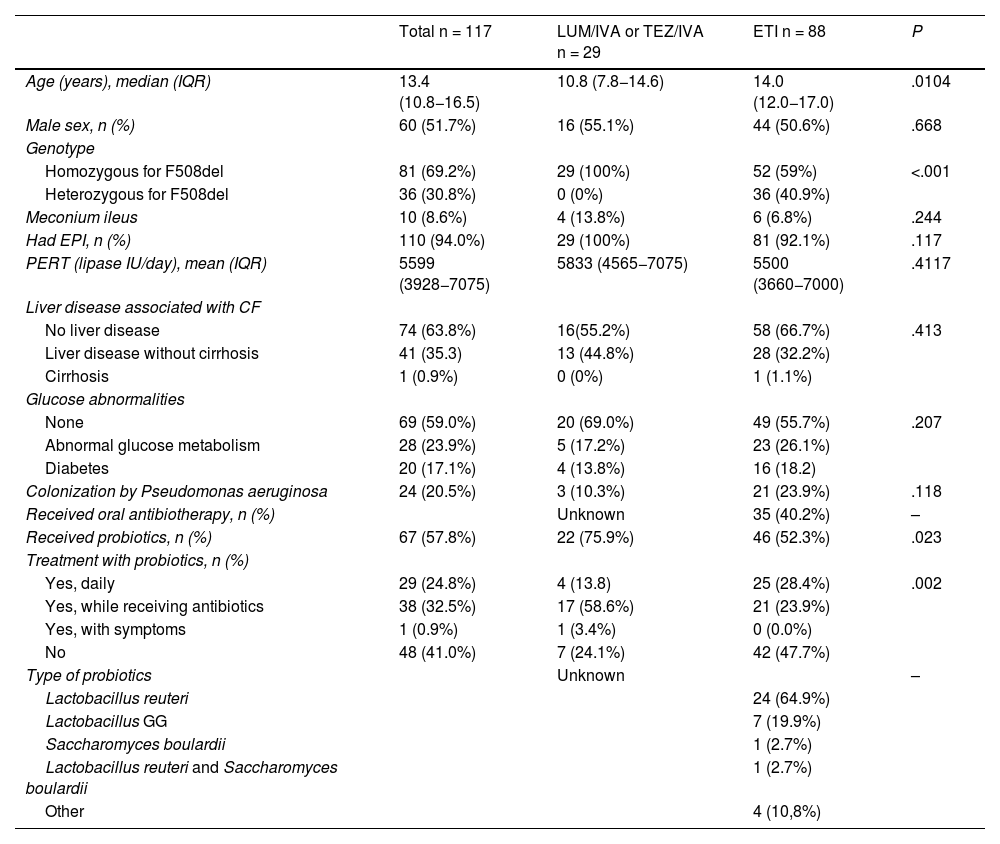
ResultsThe sample included 117 patients, with a median age of 15.5 years (95% CI, 14.0–17.0) and ages ranging from 3.2 to 47.1 years, with no differences based on sex, all of whom carried the F508del variant in at least one allele (69% homozygous). Table 1 presents the demographic and baseline clinical characteristics of the sample. These characteristics were similar in the two groups under study with the exception of age, the genotype and the detection of Pseudomonas aeruginosa.
Demographic and baseline characteristics of study participants.
| Total n = 117 | LUM/IVA or TEZ/IVA n = 29 | ETI n = 88 | P | |
|---|---|---|---|---|
| Age (years), median (IQR) | 13.4 (10.8−16.5) | 10.8 (7.8−14.6) | 14.0 (12.0−17.0) | .0104 |
| Male sex, n (%) | 60 (51.7%) | 16 (55.1%) | 44 (50.6%) | .668 |
| Genotype | ||||
| Homozygous for F508del | 81 (69.2%) | 29 (100%) | 52 (59%) | <.001 |
| Heterozygous for F508del | 36 (30.8%) | 0 (0%) | 36 (40.9%) | |
| Meconium ileus | 10 (8.6%) | 4 (13.8%) | 6 (6.8%) | .244 |
| Had EPI, n (%) | 110 (94.0%) | 29 (100%) | 81 (92.1%) | .117 |
| PERT (lipase IU/day), mean (IQR) | 5599 (3928−7075) | 5833 (4565−7075) | 5500 (3660−7000) | .4117 |
| Liver disease associated with CF | ||||
| No liver disease | 74 (63.8%) | 16(55.2%) | 58 (66.7%) | .413 |
| Liver disease without cirrhosis | 41 (35.3) | 13 (44.8%) | 28 (32.2%) | |
| Cirrhosis | 1 (0.9%) | 0 (0%) | 1 (1.1%) | |
| Glucose abnormalities | ||||
| None | 69 (59.0%) | 20 (69.0%) | 49 (55.7%) | .207 |
| Abnormal glucose metabolism | 28 (23.9%) | 5 (17.2%) | 23 (26.1%) | |
| Diabetes | 20 (17.1%) | 4 (13.8%) | 16 (18.2) | |
| Colonization by Pseudomonas aeruginosa | 24 (20.5%) | 3 (10.3%) | 21 (23.9%) | .118 |
| Received oral antibiotherapy, n (%) | Unknown | 35 (40.2%) | – | |
| Received probiotics, n (%) | 67 (57.8%) | 22 (75.9%) | 46 (52.3%) | .023 |
| Treatment with probiotics, n (%) | ||||
| Yes, daily | 29 (24.8%) | 4 (13.8) | 25 (28.4%) | .002 |
| Yes, while receiving antibiotics | 38 (32.5%) | 17 (58.6%) | 21 (23.9%) | |
| Yes, with symptoms | 1 (0.9%) | 1 (3.4%) | 0 (0.0%) | |
| No | 48 (41.0%) | 7 (24.1%) | 42 (47.7%) | |
| Type of probiotics | Unknown | – | ||
| Lactobacillus reuteri | 24 (64.9%) | |||
| Lactobacillus GG | 7 (19.9%) | |||
| Saccharomyces boulardii | 1 (2.7%) | |||
| Lactobacillus reuteri and Saccharomyces boulardii | 1 (2.7%) | |||
| Other | 4 (10,8%) | |||
Abbreviations: CF, cystic fibrosis; EPI, exocrine pancreatic insufficiency; ETI, elexacaftor/tezacaftor/ivacaftor; LUM/IVA, lumacaftor/ivacaftor; PERT, pancreatic enzyme replacement therapy; TEZ/IVA, tezacaftor/ivacaftor.
At baseline, the FC values ranged from 1 to 1450 μg/g (median, 49 μg/g; IQR, 23–108). When we analyzed baseline FC values in relation to different variables, we only found differences based on age using the nonparametric Mann-Whitney test (Fig. 1), with detection of higher values in the group aged more than 14 years. We did not find differences in relation to chronic treatment with azithromycin, colonization by Pseudomonas aeruginosa, exocrine pancreatic insufficiency or probiotic use. There were no differences in FC levels at baseline between the two treatment groups (LUM/IVA or TEZ/IVA, 48 μg/g [IQR, 26–91] vs ETI group 52 μg/g [IQR, 22–127]; P = .5677). We ought to highlight that at the start of the study, nearly half the patients (n = 57 [48%]) had FC values in the abnormal range (>50 μg/g) and 11% (all in the ETI group) had values greater than 250 μg/g indicative of intestinal inflammation.

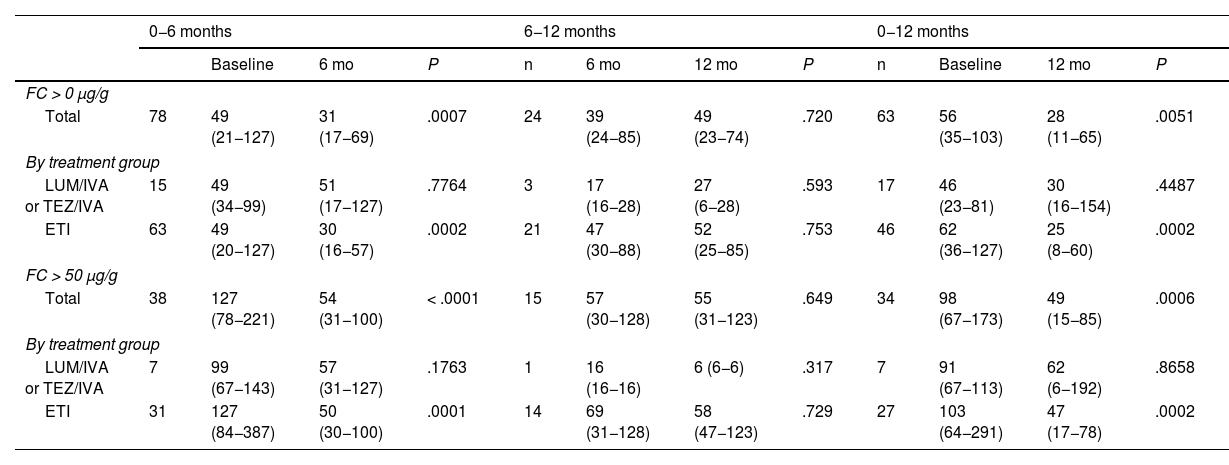
In the total sample, we found a statistically significant decrease in FC levels at 6 and 12 months of treatment relative to baseline (Table 2). We also found a progressive reduction in the proportion of patients with FC levels greater than 50 μg/g relative to baseline: at 6 months, 48.7% compared to 33.1% (P = .0067), and at 12 months, 54% compared to 33.5% (P = .0218). We did not find significant differences between 6 and 12 months of treatment. In the ETI group, only two patients at 6 months and one patient at 12 months had FC levels greater than 250 μg/g (Fig. 2).
Changes in calprotectin levels over one year of treatment with CFTR modulators.
| 0−6 months | 6−12 months | 0−12 months | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | 6 mo | P | n | 6 mo | 12 mo | P | n | Baseline | 12 mo | P | ||
| FC > 0 μg/g | ||||||||||||
| Total | 78 | 49 (21−127) | 31 (17−69) | .0007 | 24 | 39 (24−85) | 49 (23−74) | .720 | 63 | 56 (35−103) | 28 (11−65) | .0051 |
| By treatment group | ||||||||||||
| LUM/IVA or TEZ/IVA | 15 | 49 (34−99) | 51 (17−127) | .7764 | 3 | 17 (16−28) | 27 (6−28) | .593 | 17 | 46 (23−81) | 30 (16−154) | .4487 |
| ETI | 63 | 49 (20−127) | 30 (16−57) | .0002 | 21 | 47 (30−88) | 52 (25−85) | .753 | 46 | 62 (36−127) | 25 (8−60) | .0002 |
| FC > 50 μg/g | ||||||||||||
| Total | 38 | 127 (78−221) | 54 (31−100) | < .0001 | 15 | 57 (30−128) | 55 (31−123) | .649 | 34 | 98 (67−173) | 49 (15−85) | .0006 |
| By treatment group | ||||||||||||
| LUM/IVA or TEZ/IVA | 7 | 99 (67−143) | 57 (31−127) | .1763 | 1 | 16 (16−16) | 6 (6−6) | .317 | 7 | 91 (67−113) | 62 (6−192) | .8658 |
| ETI | 31 | 127 (84−387) | 50 (30−100) | .0001 | 14 | 69 (31−128) | 58 (47−123) | .729 | 27 | 103 (64−291) | 47 (17−78) | .0002 |
Values expressed as median and interquartile range.
Abbreviations: ETI, elexacaftor/tezacaftor/ivacaftor; FC, fecal calprotectin; LUM/IVA, lumacaftor/ivacaftor; TEZ/IVA, tezacaftor/ivacaftor.

The estimated change (95% CI) from treatment initiation to 12 months of treatment was greater in the ETI compared to the LUM/IVA or TEZ/IVA group, with a difference of −133 μg/g (−254 vs −13; P = .030). In the analysis adjusted for sex, probiotic use and isolation of Pseudomonas aeruginosa, the change was −130 μg/g (−259 vs −1; P = .049) (Fig. 2).
DiscussionPulmonary disease has been the main focus of clinical research on CF, since it is the factor most strongly associated with patient morbidity and mortality. The advent of CFTR modulators has been a significant therapeutic breakthrough, achieving significant improvements in lung function and a decrease in pulmonary exacerbations. However, few studies to date have described the changes in gastrointestinal health observed with these drugs, or, more specifically, analyzed potential improvements in intestinal inflammation by measuring FC levels.14–16
In our study, we analyzed a cohort of 117 patients with cystic fibrosis managed in 15 pediatric centers in Spain to describe observed changes in FC levels at 6 and 12 months of CFTR modulator therapy compared to baseline.
We found abnormal FC levels prior to treatment initiation in more than half of the patients, in agreement with previous studies,13,14 with higher levels in patients aged more than 14 years, a finding that could be related to the progression over time of chronic gastrointestinal and pulmonary inflammation and an altered intestinal microbiome, as reflected in the study conducted by De Freitas et al.12 We did not find an association with other factors, such as the use of probiotics, contrary to other authors that did find a decrease in FC with the use of certain probiotic strains.17,18 We also did not find a significant association with the presence of exocrine pancreatic insufficiency, contrary to the findings of the meta-analysis of Talebi et al.,19 who also described an association with lung function, body mass index and colonization by Pseudomonas aeruginosa. In this regard, our study did find a trend toward a positive association with colonization by Pseudomonas aeruginosa, although not statistically significant, possibly due to the smaller sample size in our study compared to the systematic review, so that FC may not only be a reliable noninvasive marker for the detection of gastrointestinal inflammation, but possibly also correlate to disease severity in patients with cystic fibrosis.
The anti-inflammatory effects at the level of the bowel already started to be described with the use of IVA as monotherapy, which was the first CFRT modulator authorized for treatment in patients with a class III genotype, in two case series that included 16 and 64 patients, respectively.20,21 Later on, following the marketing authorization of other CFTR modulators, a study assessed changes in intestinal inflammation following initiation of treatment with LUM/IVA in adolescents with cystic fibrosis followed up for one year. The median concentrations of FC decreased significantly after treatment initiation (102 μg/g [IQR, 69–210] vs713 μg/g [IQR 148–852]; P = .001).22 A recent open-label study assessed the effect of LUM/IVA in 44 children aged 1–2 years, finding a reduction in FC of –106 mg/kg at 24 weeks,23 the youngest population treated to date. However, an even more recent trial with TEZ/IVA with measurement of FC, assessment of changes in the intestinal microbiome and use of gut symptom questionnaires did not find significant differences, as was the case in our study.24
A salient finding in our study was the statistically significant difference in the decrease in FC levels when we compared treatment with dual combinations (LUM/IVA or TEZ/IVA) versus triple therapy with ETI, with the latter achieving a greater reduction in FC at both 6 and 12 months (−133). The results of triple therapy observed in our study were even better than those obtained in PROMISE, a prospective study conducted in 56 centers that found a mean difference of −66.2 μg/g (95% CI, −86.1 to −46.2) at 6 months of treatment in patients aged more than 12 years,25 and other pediatric studies, like the one conducted by Reasoner et al.,26 who found a mean decrease of −64 μg/g at 12 months (95% CI, −16 to −112).26 To date, ours is the only study that has assessed the improvement in FC levels following treatment with ETI compared to other CFTR modulator therapies, such as LUM/IVA or TEX/IVA.
There are limitations to our study, given its observational nature and the lack of randomization. However, it must be taken into account that the safety and efficacy of CFTR modulator therapy has already been proven in clinical trials, so it would be unethical to undertake a randomized study or to deny patients with eligible variants access to these drugs. On the other hand, the fact that the measurement of FC levels was not centralized may have affected the precision of the calculated magnitude of the changes. In order to minimize the impact of these limitations, we proposed comparing the results in patients treated with a dual versus triple combination in the study protocol, as well as adjusting results for factors known to affect intestinal inflammation in the statistical analysis, such as the use of probiotics or the isolation of Pseudomonas aeruginosa. We also assessed changes in FC in terms of the estimated change in each patient, as there could be differences based on the laboratory that conducted the tests.
Among the strengths of our study, we ought to highlight the prospective and multicenter design, in addition to being the largest case series published to date and the first study in Spain to assess the effectiveness of these drugs in reducing intestinal inflammation in real-world practice.
ConclusionThe use of ETI was associated with a significant reduction in FC levels compared to dual-combination therapy with LUM/IVA or TEZ/IVA, demonstrating that triple therapy is not only effective in improving respiratory parameters, but also gastrointestinal parameters such as intestinal inflammation.
FundingThis research did not receive any external funding.
Supplemental contentThe supplemental content for this article can be consulted in its online version, available at doi:10.1016/j.anpedi.2025.503836.
Saioa Vicente Santamaría, Cystic Fibrosis Unit, Department of Pediatrics, Hospital Universitario Ramón y Cajal (Madrid).
Ana Tabares González, Cystic Fibrosis Unit, Department of Pediatrics, Hospital Universitario Ramón y Cajal (Madrid).
José Ramón Gutiérrez Martínez, Cystic Fibrosis Unit, Hospital Universitario Central de Asturias (Oviedo).
Sara Sierra San Nicolás, Pediatric Gastroenterology, Hepatology and Nutrition Unit, Hospital Universitario de Canarias (Tenerife).
Pilar Ortiz Pérez, Pediatric Gastroenterology, Hepatology and Nutrition Unit, Hospital Regional Universitario de Málaga (Malaga).
Luis Peña Quintana, Department of Pediatrics, Complejo Hospitalario Insular Materno- Infantil de Las Palmas (Las Palmas de Gran Canaria).
Presentación en el XXX Congreso de la SEGHNP (Sociedad Española de Gastroenterología, Hepatología y Nutrición Pediátrica), celebrado del 25 al 27 de abril 2024. Bilbao. Comunicación oral a mesa de gastroenterología. Siendo el premio a la mejor comunicación del congreso.
Meeting presentation: this study was presented at the 30th Congress of the Sociedad Española de Gastroenterología, Hepatología y Nutrición Pediátrica (SEGHNP); April 25–27, 2024; Bilbao, Spain. It was presented as an oral communication at the Gastroenterology round table and received the award to the best communication in the congress.
Los miembros del Grupo de trabajo de Fibrosis Quística y páncreas de la SEGHNP se presentan en Anexo 1.

Anales de Pediatría (English Edition) follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals