

Karyotypes 48,XXXY and 49,XXXXY are rare sex chromosome aneuploidies that have traditionally been considered variants of Klinefelter syndrome (SK) (47,XXY), as they share features such as testicular dysgenesis and hypergonadotropic hypogonadism. However, the increased frequency of endocrine, skeletal and neuropsychological abnormalities distinguishes them from 47,XXY.1
We present the case of a boy aged 9 years and 6 months with 48,XXXY/49,XXXXY mosaicism diagnosed at 4 months of age in the context of generalised hypotonia and psychomotor delay.
The parents were healthy and not consanguineous. The findings of history taking were: term delivery (38+2 weeks) with a birth weight of 2860g (z-score, −0.71), length of 47cm (z-score, −1.12) and head circumference of 35cm (z-score, +0.09). Apgar score of 7/9. Normal genitals. Metabolic screen and hearing test with normal results. Follow-up in the cardiology department due to patent foramen oval permeable and multiple apical ventricular septal defects that resolved spontaneously. Dental caries and recurrent dental abscesses. Severe psychomotor impairment, with an intellectual quotient of less than 50, with development of smiling at 1 month of life, ability to hold head at 6 months, independent sitting at 2 years, and independent ambulation at 3 years. Scarce and repetitive speech with dyslalia, greater impairment in expressive than in receptive communication. Normal growth velocity maintained at the 50th–75th percentile for height and 90th–97th percentile for weight, bone age delay of approximately 1 year.
The findings of physical examination were: weight of 37.8kg (z-score, +2.77), height of 136.6cm (z-score, +0.23), BMI of 20.3kg/m2 (z-score, +1.44), head circumference of 50cm (z-score, −2.21) and arm span of 129.5cm. Target height 167.3±5cm (z-score, −1.36). Ptosis, epicanthal folds, upslanting palpebral fissures, broad nasal bridge, prominent ears, prognathism, multiple dental cavities, microcephaly and low anterior hairline. Bilateral limited forearm pronosupination with inability to get past the neutral position. Shortening of fifth metacarpal. Genu valgus, flat feet with superimposition of the fifth toe. Normal heart and lung sounds on auscultation. Tanner I (G1, P1, Aa), testicular volume of 1mL, penis measuring 3.4cm×1.4cm (z-score, −2.9), absence of gynecomastia.
The findings of diagnostic tests were: normal levels of thyroid hormones, follicle-stimulating hormone, luteinizing hormone, testosterone, estradiol, inhibin B and anti-Müllerian hormone. Normal carbohydrate metabolism and lipid panel. Proximal radioulnar synostosis on forearm X-ray (Fig. 1). White matter lesions of a predominantly frontal location and dilatation of periventricular Virchow-Robin spaces on magnetic resonance imaging, compatible with nonspecific leukoencephalopathy. Karyotype mosaicism with 49,XXXXY in 90% of cells and 48,XXXY in 10% of cells.
 Frontal and lateral view of the patient. (C) Forearm A-ray revealing radioulnar synostosis.")
The incidence of the 48,XXXY and 49,XXXXY variants is considerably lower compared to KS, of 1 in 17000 to 1 in 50000 live births and 1 in 85000 to 1 in 100000 live births, respectively.1,2 We present a case of an exceedingly rare form of mosaicism, 48,XXXY/49,XXXXY, whose incidence is unknown. While KS is associated with high stature, as does the 48,XXXY variant, the 49,XXXXY variant presents with short stature for reasons that have yet to be established. Our patient had normal stature, although his height was high relative to his target height. Most of the features described above are shared by both variants and 47,XXY variant2 (Table 1). The frequency of radioulnar synostosis increases with increasing number of X chromosomes.2 Thus, although these features have been described in 47,XXY/46,XX mosaicism, they are more frequent in variants with tetrasomy and pentasomy.3 Other congenital malformations, such as cleft palate, kidney or hip dysplasia or inguinal hernia, were not present in our patient, despite their increased prevalence and severity in individuals with the 49,XXXXY variant.1,4 Hypogonadism may manifest with micropenis, cryptorchidism, scrotal hypoplasia or hypergonadotropic hypogonadism. The latter is a feature shared with KS and its variants. In the prepubertal stage, our patient had a testicular volume of 1mL and micropenis, which are also common findings in all variants.1 The 47,XXY and the tetrasomy and pentasomy variants manifest with a broad spectrum of cognitive impairment, with the most severe impairment found in association with the 49,XXXXY variant1,4 (Table 1). Our patient had severe psychomotor impairment with significant deficits in expressive language, which is a common feature of all variants. Tartaglia et al. described the presence of motor delays and hypotonia in 100% of 49,XXXXY individuals, with development of independent ambulation at a mean age of 25.5 months.1 Visootsak et al. estimated that the intellectual quotient decreases by 10–15 points with each additional X chromosome.2,5 Abnormalities of the brain parenchyma (ventriculomegaly, white matter hyperintensities) and the craniocervical junction are also associated to variants of KS, as described by Milani et al.6 With the exception of the nonspecific white matter lesions, we did not find any of these abnormalities in our patient.
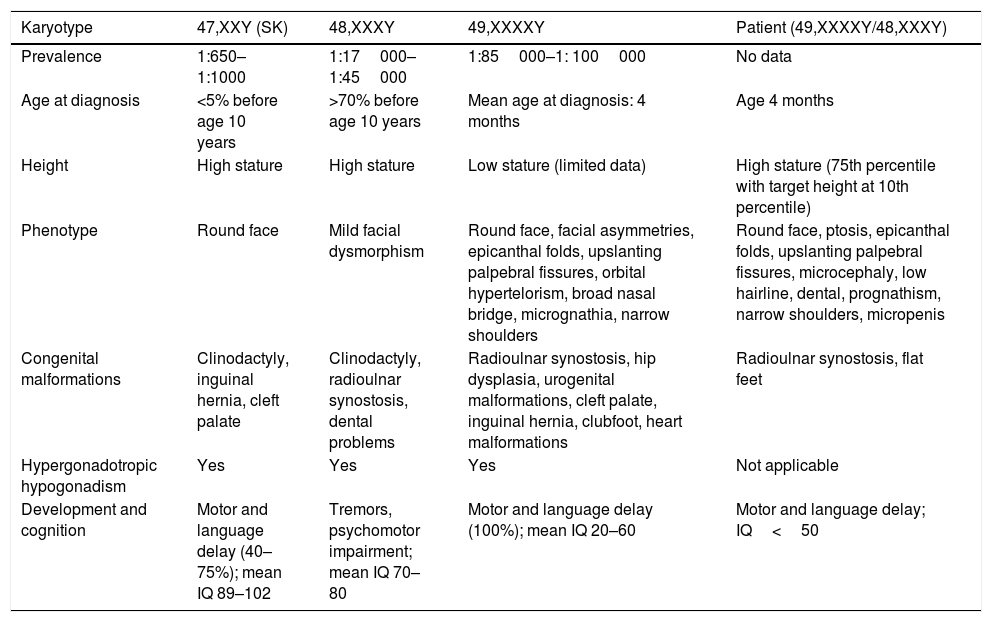
Clinical characteristics of chromosome variants 47,XXY; 48,XXXY; 49,XXXXY and of the patient with 48,XXXY/49,XXXXY mosaicism.
| Karyotype | 47,XXY (SK) | 48,XXXY | 49,XXXXY | Patient (49,XXXXY/48,XXXY) |
|---|---|---|---|---|
| Prevalence | 1:650–1:1000 | 1:17000–1:45000 | 1:85000–1: 100000 | No data |
| Age at diagnosis | <5% before age 10 years | >70% before age 10 years | Mean age at diagnosis: 4 months | Age 4 months |
| Height | High stature | High stature | Low stature (limited data) | High stature (75th percentile with target height at 10th percentile) |
| Phenotype | Round face | Mild facial dysmorphism | Round face, facial asymmetries, epicanthal folds, upslanting palpebral fissures, orbital hypertelorism, broad nasal bridge, micrognathia, narrow shoulders | Round face, ptosis, epicanthal folds, upslanting palpebral fissures, microcephaly, low hairline, dental, prognathism, narrow shoulders, micropenis |
| Congenital malformations | Clinodactyly, inguinal hernia, cleft palate | Clinodactyly, radioulnar synostosis, dental problems | Radioulnar synostosis, hip dysplasia, urogenital malformations, cleft palate, inguinal hernia, clubfoot, heart malformations | Radioulnar synostosis, flat feet |
| Hypergonadotropic hypogonadism | Yes | Yes | Yes | Not applicable |
| Development and cognition | Motor and language delay (40–75%); mean IQ 89–102 | Tremors, psychomotor impairment; mean IQ 70–80 | Motor and language delay (100%); mean IQ 20–60 | Motor and language delay; IQ<50 |
IQ, intellectual quotient.
In conclusion, karyotype 48,XXXY/49,XXXXY is a rare sex chromosome aneuploidy that, while sharing some features with KS, is characterised by a higher frequency of congenital anomalies (especially proximal radioulnar synostosis) and cognitive and behavioural abnormalities that are generally more severe than those found in association with the 47,XXY variant. These patients require multidisciplinary follow-up.
Please cite this article as: del Río RG, dos Santos TJ, Travieso-Suárez L, Muñoz Calvo MT, Argente J. Cariotipo 48,XXXY/49,XXXXY y sinóstosis radioulnar proximal. An Pediatr (Barc). 2018;88:282–284.
