









Disorders of sex development (DSD) include a wide range of anomalies among the chromosomal, gonadal, and phenotypic (genital) characteristics that define sexual differentiation. At present, a definition as Different sexual development (DSD) is currently preferred. They originate in the pre-natal stage and are classified according to the sex chromosomes present in the karyotype. The known genetic causes are numerous and heterogeneous, although, in some cases, they may be secondary to maternal factors and/or exposure to endocrine-disrupting chemicals (EDCs). The diagnosis and treatment of DSD always requires multidisciplinary medical and psychosocial care. An aetiological diagnosis needs the interaction of clinical, biochemical (hormonal), genetic, imaging and, sometimes, surgical examinations. The treatment should deal with sex assignment, the possible need for hormone replacement therapy (adrenal if adrenal function is impaired, and with sex steroids from pubertal age if gonadal function is impaired), as well as the need for surgery on genital structures (currently deferred when possible) and/or on gonads (depending on the risk of malignancy), the need of psychosocial support and, finally, an adequate organisation of the transition to adult medical specialties. Patient Support Groups have a fundamental role in the support of families, as well as the interaction with professional and social media. The use of Registries and the collaboration between professionals in Working Groups of national and international medical societies are crucial for improving the diagnostic and therapeutic tools required for the care of patients with DSD.
Las anomalías de la diferenciación sexual (ADS) engloban un amplio espectro de discordancias entre los criterios cromosómico, gonadal y fenotípico (genital) que definen la diferenciación sexual; actualmente, se aboga por la denominación de «desarrollo sexual diferente» (DSD). Su origen es congénito; se clasifican en función de los cromosomas sexuales presentes en el cariotipo; las causas genéticas conocidas son muy diversas y heterogéneas, aunque algunos casos pueden ser secundarios a factores maternos o medioambientales. Su diagnóstico y tratamiento requieren siempre una atención médica y psicosocial multidisciplinar. El diagnóstico etiológico precisa la interacción entre las exploraciones clínicas, bioquímicas (hormonales), genéticas, de imagen y, eventualmente, quirúrgicas. El tratamiento debe abordar la asignación de género, la posible necesidad de tratamiento hormonal substitutivo (suprarrenal si hay insuficiencia suprarrenal y con esteroides sexuales si hay insuficiencia gonadal a partir de la edad puberal), la necesidad de intervenciones quirúrgicas sobre las estructuras genitales (actualmente se tiende a diferirlas) y/o sobre las gónadas (en función de los riesgos de malignización), la necesidad de apoyo psicosocial y, finalmente, una adecuada programación de la transición a la atención médica en las especialidades de adultos. Las asociaciones de personas afectadas tienen un papel fundamental en el apoyo a familias y la interacción con los medios profesionales y sociales. La utilización de Registros y la colaboración entre profesionales en Grupos de Trabajo de sociedades médicas nacionales e internacionales es fundamental para avanzar en mejorar los medios diagnósticos y terapéuticos que precisan los DSD.
Disorders of sex development is a term that encompasses a broad spectrum of conditions with atypical development of the chromosomal, gonadal and phenotypic (genital) characteristics that define sexual differentiation.1 The 2006 Chicago Consensus Statement1,2 referred to these conditions as “disorders or abnormalities of sex development”, although it still also used the term “intersex” (Table 1). The growing awareness of the disapproval elicited by this new medical terminology has led to a progressive reconsideration of the terms, and the term currently proposed to refer to these conditions is “differences of sex development” (DSD).3
Sexual differentiation during prenatal development involves a series of processes whose initiation and regulation involve numerous genes, proteins and hormones. The first stage in gonadal and genital development is shared by both sexes and spans the first 6 weeks following conception, an interval during which the embryo is pluripotent. Gonadal differentiation starts in the 7th week and is regulated by a multitude of genes, with the SRY gene in the Y chromosome playing a key role in the development of the testes. Genital differentiation (internal and external) is regulated by the effects of hormones synthesised by the testes in male embryos, or by their absence in female embryos. Any abnormality of genetic or environmental origin that impacts these processes at any level may result in inadequate development of the gonads (gonadal dysgenesis), the internal genitalia (absence or abnormal features) and/or the external genitalia (insufficient or excessive virilization). These abnormalities may be apparent at birth, manifesting as genital ambiguity or discordance between genotypic and phenotypic sex, during puberty, manifesting as delayed puberty, amenorrhoea or insufficient or excessive virilization, or later in life, manifesting as infertility or early menopause, and it is important to remember that they can be associated to anomalies in other systems or be life-threatening if they are associated with adrenal insufficiency. The approach to their management is also critical in infants when it comes to gender assignment. For all the above reasons, these conditions always require medical and psychosocial care delivered by a multidisciplinary team.
With the exception of hypospadias as an isolated genital malformation, the nonclassic form of congenital adrenal hyperplasia (CAH) and sex chromosome disorders, their incidence is less than 1/2000 individuals, so they are considered “rare diseases”.
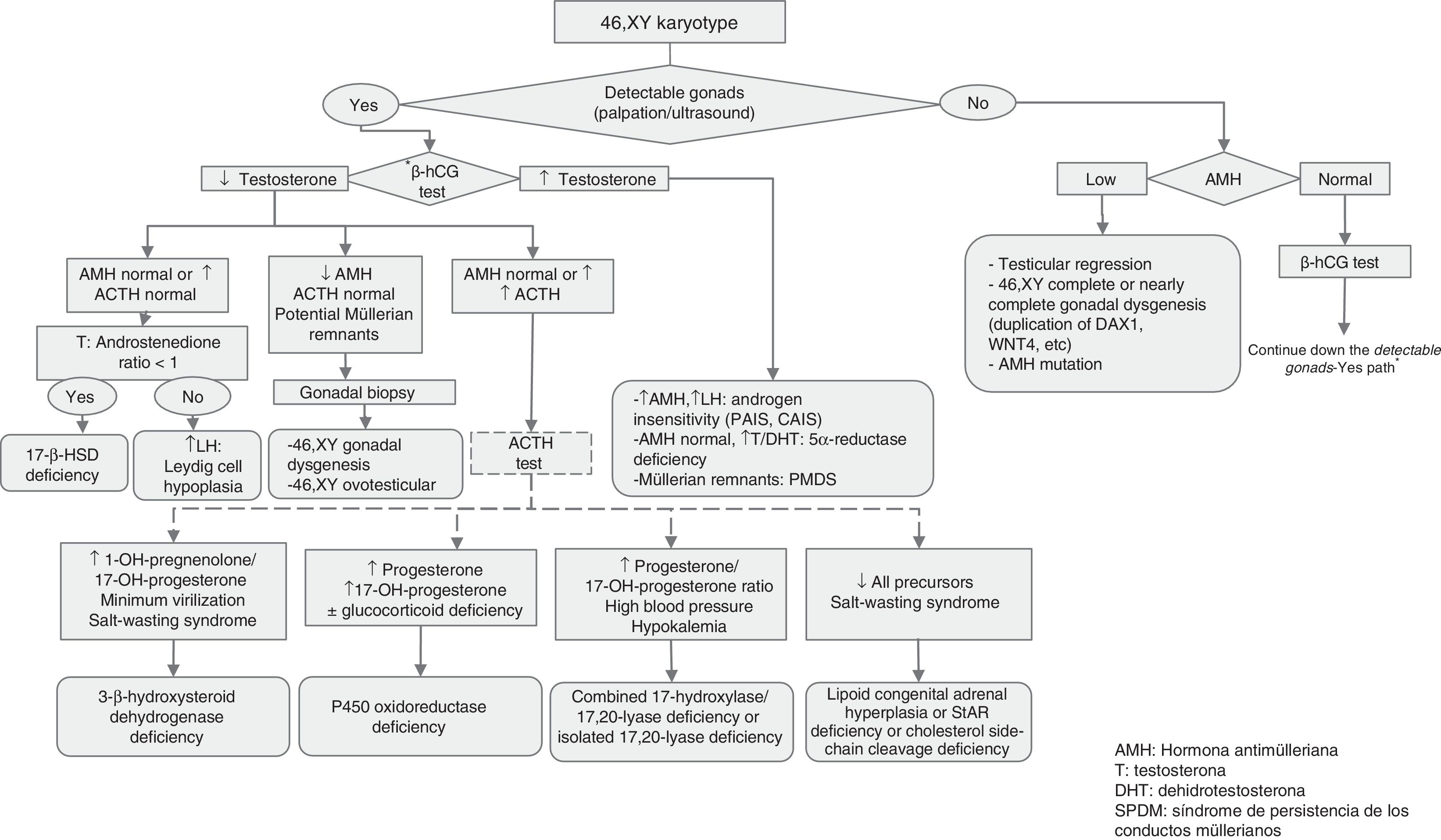
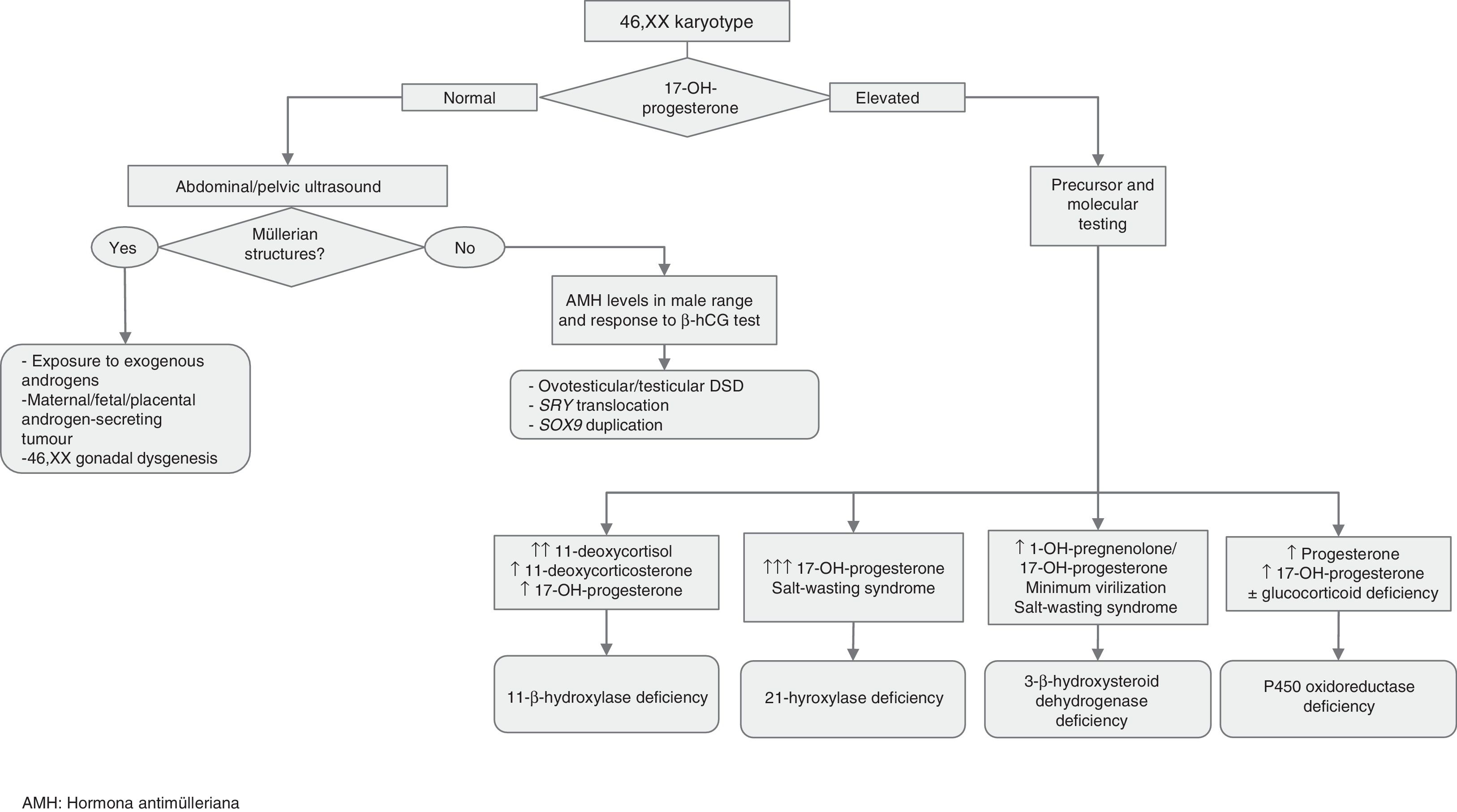
Approach to the diagnosis of genital ambiguity or suspected DSD (Figs. 1 and 2)In addition to genital ambiguity in the neonatal period, other clinical manifestations suggestive of the presence of a DSD are:
- 1.
Male genitalia with proximal (scrotal) hypospadias, micropenis, bilateral cryptorchidism, testicular atrophy and/or distal or medial hypospadias associated with unilateral cryptorchidism.
- 2.
Female genitalia with inguinal or labial mass and/or enlarged clitoris with posterior labial fusion.
The recommended approach to the diagnosis of any suspected DSD now follows, although some of the diagnostic tests are used exclusively in the assessment of newborns with genital ambiguity. As noted above, the diagnosis should be made by a multidisciplinary team.
History taking and physical examinationPersonal history- •
Parental consanguinity.
- •
Potential prenatal exposure to androgens, androgen antagonists or other drugs.
- •
Maternal virilization during pregnancy.
Family history of hypospadias, infertility, amenorrhoea or early menopause, salt-losing or unexplained infant deaths.
Physical examinationA careful inspection with palpation of the genitalia must be performed:
- •
Assess the degree of virilization/masculinisation: there are two scales that can be used interchangeably.
- –
In female individuals (XX), although it can also be useful in general for any other chromosome pair or when the results of karyotyping are not yet available, the Prader staging system, which can be found in p. 9 of the document Guía de actuación en las anomalías de la diferenciación sexual (ADS)/desarrollo sexual diferente (DSD)4 (“Guideline for the management of disorders/differences of sex development (DSD)”), to which we will refer to from hereon as the DSD Guideline.
- –
In male individuals (XY), by means of the external masculinisation score (p. 10 of the DSD Guideline).4
- •
Palpation of the gonads along the inguinal canal (from the labioscrotal fold to the abdomen).
- •
Assessment of hydration and blood pressure. In cases with persistent jaundice accompanied by recurrent episodes of hypoglycaemia, the possibility of hypopituitarism with growth hormone and cortisol deficiency in addition to gonadotropin deficiency should be considered.
- •
Rule out additional dysmorphic features, as genital malformations may occur in the context of multiple malformation syndromes.
There are three fundamental tests:
Karyotype analysis (peripheral blood)Essential during the diagnostic evaluation, as it helps place the patient in one of the 3 main categories of the DSD classification (Table 1). It can be supplemented with FISH analysis using probes specific for the Y (SRY) and X (DX1) chromosomes.
Hormone evaluation from 48h post birth5- •
17-Hydroxyprogesterone. It can be used to screen for CAH secondary to 21-hydroxylase deficiency, which is the most common cause of 46,XX DSD. Its levels must be measured in all newborns with bilateral cryptorchidism or with genital ambiguity.6
- •
Dehydroepiandrosterone (DHEA), progesterone and, if possible, 17-hydroxypregnenolone and 11-deoxycortisol. They allow the diagnosis of less common forms of CAH and various inherited metabolic disorders.
- •
Testosterone, follicle-stimulating hormone (FSH) and luteinising hormone (LH). Measurement of the basal serum levels of testosterone and its precursors in patients with 46,XY DSD is useful in the first year of life, performing tests within 36h of birth and during mini-puberty, between days 15 and 90 post birth (a window that can be expanded to 6 months of age, during the descent in testosterone levels). The levels of LH and FSH should also be measured in the sample obtained for the second test.
- •
Basal cortisol and adrenocorticotropic hormone (ACTH). Essential for diagnosis of panhypopituitarism and enzymatic disorders that affect adrenal steroidogenesis.
- •
Anti-Müllerian hormone (AMH) and inhibin B. Measurement of serum levels of AMH and/or inhibin B is used to assess Sertoli cell function.7,8
- •
Urinary steroid profile: the ratio of the measured urine concentrations of precursor metabolites and the steroid products of the enzyme with the potential defect may be a more specific and sensitive method to detect the defect compared to measurement of levels in blood.9
It is important to assess for the presence of the gonads, uterus and/or vagina. The identification of these structures, especially of the gonads, is not always easy, and therefore lack of detection does not necessarily entail their absence.
Second-line diagnostic testsThese are used when the aetiology remains unclear, or further assessment is desired in cases of suspected DSD:
- •
Beta-human chorionic gonadotropin (β-hCG) simulation test (short duration): for assessment of testicular function based on the response of Leydig cells, measuring the levels of synthesised testosterone and its precursors and metabolites, such as dihydrotestosterone.7,10
- •
Indications, protocol and interpretation detailed in pp. 13–14 of the DSD Guideline.4
- •
ACTH test:
- •
Indications and protocol detailed in p 14 of the DSD Guideline.4
- •
Additional imaging tests as recommended by the surgeon or urologist, such as a genitogram, retrograde urethrogram or cystoscopy/vaginoscopy. Magnetic resonance imaging is indicated as an alternative to laparoscopic examination when the gonads are not detected by ultrasound.
- •
Laparoscopic examination with gonadal biopsy may be needed for assessment of potential testicular dysgenesis in XY males.
- •
Molecular studies: the results of karyotype analysis, history, clinical manifestations and blood and imaging tests may guide the aetiological diagnosis. If testing detects abnormalities in the sex chromosomes, the aetiology becomes clear. However, when the patient has a 46,XX or 46,XY karyotype, the overall findings may suggest a monogenic disorder, in which case the logical next step would be to analyse the candidate gene. The diagnostic algorithms of Figs. 1 and 2 combined with Tables 1–4 can be used to follow this approach to diagnosis.
Table 1.Classification of differences/disorders of sex development (DSD).
Sex chromosome DSD 47,XXY: Klinefelter syndrome and variants 45,X0 and 45,X0/46,XX mosaicism: Turner syndrome and variants 45,X0/46,XY: mixed gonadal dysgenesis 46,XX/46,XY: chimeric/ovotesticular DSD 47,XYY 46,XY DSD Disorders of gonadal development 46,XY gonadal dysgenesis (complete or partial) (SRY, SOX9, NR5A1, WT1, DHH, etc.) 46,XY ovotesticular DSD Testicular regression syndrome (includes anorchidism and vanishing testis syndrome) Abnormal genital development due to disorders in hormone synthesis or action Disorders in androgen synthesis LH receptor defects (Leydig cell hypoplasia or aplasia; LHCGR) Smith-Lemli-Opitz syndrome (7-dehydrocholesterol reductase deficiency: DHCR7) Testosterone biosynthesis defect Lipoid congenital adrenal hyperplasia (StAR) Cholesterol side-chain cleavage enzyme deficiency (CYP11A1) 3-β-Hydroxysteroid dehydrogenase deficiency (HSD3B2) 17α-Hydroxylase/17,20 lyase deficiency (CYP17A1) Cytochrome P450 oxidoreductase deficiency (POR) Cytochrome b5 reductase deficiency (CYB5) 17β-Hydroxysteroid dehydrogenase deficiency (HDS17B3) 5α-Reductase 2 deficiency (SRD5A2) Disorders in androgen action Androgen insensitivity syndrome (AIS; complete or partial [CAIS or PAIS]) Drugs and environmental modulators Disorders in anti-Müllerian hormone synthesis or action Persistent Müllerian duct syndrome (AMH/AMHR2) Other Syndromic associations of male genital development (e.g. cloacal anomalies, Aarskog syndrome, Robinow syndrome, etc.) Severe early onset intra-uterine growth restriction Isolated hypospadias (CXorf6 or MAMLD1) Congenital hypogonadotropic hypogonadism Cryptorchidism (INSL3, RXFP2 [or INSL3R or GREAT]) 46,XX DSD Disorders of gonadal development 46,XX gonadal dysgenesis 46,XX ovotesticular DSD 46,XX testicular DSD (SRY, dup SOX9, RSPO1) or 46,XX male syndrome/sex reversal Disorders in genital development due to androgen excess Foetal origin 21-Hydroxylase deficiency (CPY21A2) 11-β-Hydroxylase deficiency (CYP11B1) Cytochrome P450 oxidoreductase deficiency (POR) Cytochrome b5 reductase deficiency (CYB5 3-β-Hydroxysteroid dehydrogenase deficiency (HSD3B2) Glucocorticoid receptor mutations (NR3C1) Fetoplacental origin Placental and foetal aromatase deficiency (CYP19A1) Cytochrome P450 oxidoreductase deficiency (POR) Foetal or maternal androgen-secreting tumours Maternal origin Androgenic drugs Maternal virilising tumours (e.g. luteoma, Krukenberg tumour) Other Malformation syndromes Müllerian agenesis/hypoplasia (Rokitansky-Hauser syndrome type I and type II-MURCS) Uterine anomalies (e.g. MODY 5) Vaginal atresia Labial adhesions Source: Lee et al.2
Table 2.Sex chromosome DSD.
Gonads Internal genitalia External genitalia Laboratory abnormalities Management Wolffian structures Müllerian structures 1. Disorders with genital ambiguity 45,X0/46,XY mosaicism (mixed gonadal dysgenesis) Dysgenetic testes or streak gonads (normal in some cases) Hypoplastic, in some cases unilateral (present in the pelvic side where there is testicular tissue: homolateral production of AMH) Hypoplastic, in some cases unilateral (present in the pelvic side where there is no testicular tissue: homolateral absence of AMH production) Variable: partial virilization (rarely fully male or female). Suspect in case of asymmetrical external genitalia (including unilateral cryptorchidism). May be associated with short stature and, less frequently, heart or kidney anomalies. Hypergonadotropic hypogonadism (testosterone and AMH levels depend on the presence on normal testicular tissue) Confirmation of diagnosis: gonadal biopsy→dysgenetic gonad: high risk of gonadoblastoma (gonadectomy) 46,XX/46,XY mosaicism (33% cases of ovotesticular DSD or chimerism) Testicle and ovary in a single individual or mixture of ovarian and testicular tissue in the same gonad (ovotestis) Variable (present in the pelvic side where there is testicular tissue: homolateral production of AMH) Variable (present in the pelvic side where there is no testicular tissue) Variable: partial virilization (rarely completely male or female) Confirmation of diagnosis: gonadal biopsy→Ovotestis: risk of malignant transformation, 3% (consider resection of testicular tissue) 2. Disorders without genital ambiguity Turner syndrome and variants (45,X0 and 45,X0/46,XX mosaicism). Gonadal dysgenesis (90%) Absent Present (with variable degree of development) Female with stigmata: short stature, pterigium colli, hypoplastic wide set breasts, etc. Hypergonadotropic hypogonadism depending on degree of dysgenesis – Recombinant GH treatment if short stature
– Most cases: sex steroid therapy for induction of puberty and replacementKlinefelter syndrome and variants (47,XXY, etc.) Hyalinized testes (small testes) Present Absent Male. May be associated with tall stature, gynecomastia and features secondary to progressive androgen deficiency. Infertility Hypergonadotropic hypogonadism (greater elevation of FSH compared to LH due to involvement mainly of testicular Sertoli cells) – May require testosterone replacement therapy based on LH (↑↑) and testosterone (↓) levels
– If significant gynecomastia: consider surgery47,XYY DSD Normal testes in most cases Present Absent Male Normal testicular function in most cases Usually have height greater than family height and mildly impaired cognitive development. Usually detected on karyotype analysis performed for reasons unrelated to gonadal function Table 3.46,XY DSD.
Gonads Internal genitalia External genitalia Laboratory abnormalities Management Wolffian structures Müllerian structures 1. Disorders of gonadal development (testicular dysgenesis) 46XY partial or complete gonadal dysgenesis (Swyer syndrome) Present (dysgenetic) – Partial form: present (hypoplastic)
– Complete form: absent– Partial form: absent
– Complete form: present. Depending on mutation, may present with different phenotypesa– Partial form: partial virilization (variable)
– Complete form: female↓Testosterone
(basal and/or β-hCG test)
↓ AMH– Imaging test (ultrasound): gonadal position
– Confirmation of diagnosis: gonadal biopsy and molecular study of gonadal sex differentiation genes (SRY, WT1, etc.a)46XY ovotesticular DSD (7% of cases of ovotesticular DSD or chimerism) Present (dysgenetic with mixed ovarian and testicular tissue in the same gonad) Present (+/− hypoplastic) Absent Variable: partial virilization (rarely completely female or male) – Imaging test (ultrasound): gonadal position
– Confirmation of diagnosis: gonadal biopsyTesticular regression syndrome (loss of testicular function/tissue in early development) Anorchidism Present (+/− hypoplastic) Absent (or present if at very early stage) Variable: partial virilization (if very early: female) – Imaging test (ultrasound): undetectable gonads Vanishing testes syndrome (loss of testicular function/tissue in late development) Anorchidism Present Absent Male – Confirmation of diagnosis: laparoscopy. Molecular study of gonadal sex differentiation genes (SRY, WT1, etc.) 2. Disorders in androgen synthesis or action 2.1. Low or absent testosterone response after β-hCG stimulation test Enzymatic disorders of testosterone synthesis 17-β-Hydroxysteroid-dehydrogenase Testicles Present (normal) Absent (blind vagina in some forms, such as StAR, cholesterol side-chain cleavage or POR deficiency and Smith-Lemli-Opitz syndrome) Female or genital ambiguity. Very rarely male
Puberty: signs of virilizationβ-hCG test:
↓Testosterone
AMH normal or ↑
ACTH normal
T/Androstenedione <1ACTH test not indicated
Confirmation of diagnosis: genetic-molecular study of HSD17B3 gene (9q22)3-β-Hydroxysteroid dehydrogenaseb Male with variable undermasculinization β-hCG test:
↓Testosterone
AMH normal or ↑
↑ACTHACTH test:
↑17-OH-pregnenolone/17-OH-progesterone
Salt-wasting syndrome
Confirmation of diagnosis: genetic-molecular study of HSD3B2 gene (1p13.1)Combined 17-α-hydroxylase/17,20-lyaseb deficiency or isolated lyase deficiency Present (normal or hypoplastic) Female or genital ambiguity (the latter is common in POR and cytochrome b5 deficiency) ACTH test:
↑ progesterone/17-OH-progesterone ratio
Hypertension
Hypokalaemia
In isolated lyase deficiency: ↑ only in progesterone and 17-OH-progesterone/androstenedione ratio
Confirmation of diagnosis: genetic-molecular study of CYP17A1 gene (10q24.3)Lipoid congenital adrenal hyperplasia (StAR protein deficiency) and cholesterol side-chain cleavage enzyme deficiencyb ACTH test:
↓ of all adrenal and gonadal steroids
Ultrasound: in StAR protein deficiency, enlarged adrenals due to lipid accumulation
Confirmation of diagnosis: genetic-molecular study of StAR (8p11.2) and CYP11A1 (15q24.1) genesP450 oxidoreductase deficiency (POR)b ACTH test:
↑ Progesterone
↑ 17-OH-progesterone
+/− glucocorticoid deficiency
Confirmation of diagnosis: genetic-molecular study of POR gene (7q11.2)Cytochrome b5 deficiency Testicles Present (normal or hypoplastic) Absent (blind vagina in some cases such as StAR, cholesterol side chain cleavage enzyme or POR deficiency and Smith-Lemli-Opitz syndrome) Female or genital ambiguity (the latter is common in POR and cytochrome b5 deficiency) β-hCG test:
↓ testosterone
AMH normal or ↑
↑ACTHMethemoglobinemia
β-hCG test: ↑ progesterone and 17-OH-progesterone/androstenedione ratio
Confirmation of diagnosis: genetic-molecular study of CYB5A gene (18q22.3)Defects in the alternative or backdoor steroid hormone synthesis pathway ACTH test: normal
β-hCG test:
Testosterone normal or ↓
DHT ↓↓
Confirmation of diagnosis: genetic-molecular study of AKR1C2 and AKR1C4 genesSmith-Lemli-Opitz syndromeb Undermasculinization with peculiar phenotype (dysmorphic face, psychomotor retardation, heart and visceral anomalies) ↑ 7-dehydrocholesterol
Confirmation of diagnosis: genetic-molecular study of DHCR7 gene (11q13.4)2.2 Normal or increased testosterone response after β-hCG test Gonads Internal genitalia External genitalia Laboratory abnormalities Management Wolffian structures Müllerian structures 5-α-Reductase type 2 Testicles Present
Hypoplastic vaginaAbsent Variable virilization Normal testosterone
Normal AMH
After β-hCG: Testosterone/DHT>30Diagnosis: genetic-molecular study of SDR5A2 gene (2p23) Androgen insensitivity (partial forms [PAIS] and complete forms [CAIS] or Morris syndrome) Testicles – Partial forms (PAIS): +/− hypoplastic
– Complete form (CAIS): absentAbsent
Atrophic uterus in exceptional cases– Partial forms (PAIS): variable
– Complete form (CAIS): female with scant pubic and axillary hairTestosterone ↑ or normal
AMH ↑ or normal
LH ↑ or normalDiagnosis: genetic-molecular study (AR; Xq12) 3. Disorders of anti-Müllerian hormone synthesis or action Persistent Müllerian duct syndrome (abnormality of the anti-Müllerian hormone [AMH] or its receptor [AMHR2]) Testicles (bilateral cryptorchidism with/without inguinal hernia) Present Present Male Normal testosterone
AMH ↓ (AMH mutation) or normal (AMHR2 mutation)Diagnosis: genetic-molecular study of AMH (19p13.3) or AMHR2 (12q13.13) gene based on AMH levels 4. Malformation syndromes (cloacal exstrophy, VACTERL/VATER, etc.), extreme intrauterine growth restriction (lack of stimulation of Leydig cells in the first 15 weeks by placental β-hCH substituting LH), etc. aPhenotypes associated with mutations: WT1 (kidney anomalies, Wilms tumour, gonadal tumours), SF1 (NR5A1: adrenal insufficiency and partial hypogonadotropic hypogonadism, only when the mutation affects both alleles), SOX9 (campomelic dysplasia), CBX2 (presence of ovaries and Müllerian remnants), DHH (minifascicular neuropathy), del 9p24.3 (DMRT1 and DMRT2: intellectual disability), del Xq13.3 (ATRX): intellectual disability, thalassemia), ARX (lissencephaly, epilepsy), TSPYL1 (sudden infant death syndrome), DAX1dup (complete or partial dysgenesis), EMX (intellectual disability, one kidney), FGFR2 (craniosynostosis), GATA4 (congenital heart defect), HHAT (short stature, chondrodysplasia, muscular hypertrophy, myopia, mild intellectual disability, MAMLD1 (hypospadias), MAP3K1, WNT4dup, WWOXdel; FOG2/ZFPM2 (congenital heart defect?).
Table 4.46,XX DSD.
Gonads Internal genitalia External genitalia Laboratory abnormalities Approach Wolffian structures Müllerian structures 1. Disorders of gonadal development (ovarian dysgenesis) 46,XX gonadal dysgenesis Present (dysgenesis) Absent Present Female Hypergonadotropic hypogonadism (↓estradiol, ↑FSH and LH) Confirmation of diagnosis: gonadal biopsy→gonadal dysgenesis, and molecular study of gonadal sex differentiation genes (duplication of SOX9, RSPO1a, WNT4, etc.b) 46,XX ovotesticular DSD (33% of ovotesticular DSD cases) Present (dysgenetic, with a mixture of ovarian and testicular tissue in the same gonad) (usually absent) Variable: present in most, with different degrees of development Variable: ambiguous, female or male Hypergonadotropic hypogonadism
AMH values in male range (based on the presence of functional testicular tissue)
β-hCG test: post-stimulation increase in testosterone (based on the presence of functional testicular tissue)Diagnosis: genetic-molecular study:
– Most cases: unknown aetiology
– SRY gene (Yp11.2) negative in most; in 10%-15% of cases: SRY translocation to X chromosome or autosome (SRY+)
– Occasionally: SOX9 duplication (17q24), mutation in RSPO1 genea (1p34.3) or WNT4 gene (1p36.12)
Confirmation of diagnosis: gonadal biopsy→Ovotestis: 3% risk of malignant transformation (consider resection of testicular tissue)46,XX testicular DSD (XX male or de la Chapelle syndrome) Testicles (testicular atrophy) Present Absent – Normal male (85% cases)
– Hypospadias or mild genital ambiguity (15% cases)Hypergonadotropic hypogonadism (↓ testosterone, ↑FSH and LH) Diagnosis: only molecular-genetic study (80% SRY gene translocation from paternal Y chromosome to maternal X chromosome (SRY+); occasionally, SOX9 duplication) 2. Androgen excess 2.1. Congenital adrenal hyperplasia (manifests with adrenal insufficiency) 21-Hydroxylase deficiency Present (ovaries) Absent or limited development Present Ambiguous genitalia (classic or simple virilising form). Rarely penis without hypospadias ↑↑17-OH-Progesterone
Classic form: salt wasting with hyponatremia, hyperkalemia and hypotension (first weeks of life)Diagnosis: molecular-genetic study of CYP21A2 gene (6p21.3) 11-β-Hydroxylase deficiency ↑17-OH-progesterone
↑11-deoxycorticosterone (mineralocorticoid activity), ↑11-deoxycortisol
Hypertension
No salt wastingDiagnosis: molecular-genetic study of CYP11B1 gene (8q21) P450 oxidoreductase deficiency Variable (from ambiguous genitals to normal female) ↑17-OH-Progesterone
↑Testosterone ↑Progesterone ↑Corticosterone
+/− glucocorticoid deficiencyDiagnosis: molecular-genetic study of POR (7q11.2) 3-β-Hydroxysteroid dehydrogenase deficiency Female external genitalia with absent or mild virilization (clitoromegaly) ↑17-OH-Progesterone
↑17-OH-Pregnenolone
Salt-wasting syndromeDiagnosis: molecular-genetic study of HSD3B2 (1p13.1) 2.2. Gestational hyperandrogenism Exposure to maternal androgens or synthetic progestogens (drugs, maternal virilising tumours, fetoplacental aromatase deficiency, foetal or placental androgen-secreting tumours) Present (ovaries) Absent or limited development Present Variable degree of virilization ↑Testosterone +/− androgen precursors Diagnosis: peripartum anamnesis and blood tests in mother and newborn
Molecular-genetic study if aromatase deficiency is suspected: CYP19A1 gene (15q21.2)2.3. Glucocorticoid receptor mutations Exposure to maternal androgens or synthetic progestogens (f drugs, maternal virilising tumours, fetoplacental aromatase deficiency, foetal or placental androgen-secreting tumours) Present (ovaries) Absent Present Variable degree of virilization ↑ACTH and ↑cortisol with signs of adrenal insufficiency
↑Androgen precursors
↑TestosteroneDiagnosis: molecular-genetic study of NR3C1 gene (5q31.3) bOther genes whose mutations are associated with 46,XX ovarian dysgenesis, ovotesticular chimerism or testicular DSD are: BMP15, FGF9dup, FOXL2 (blepharophimosis, ptosis and epicanthal folds), NR5A1 (ovarian dysgenesis, ovotesticular chimerism or testicular DSD testicular in case of Arg92Trp mutation), SOX3dup.
The different molecular studies are used to assess for a variety of genetic disorders:
- –
Detection of pathogenic changes in the sequence of a gene: the classic Sanger sequencing technique is used when there is a clear candidate gene (for instance, CYP21A2 in case of suspected 21-hydroxylase deficiency or AR in case of androgen insensitivity). However, due to the large number of candidate genes that may be involved in DSDs, many laboratories use next generation sequencing (NGS) techniques that allow the simultaneous sequencing of a variable number of genes; these methods can be used to find the genetic cause quicker and at lower cost.
- –
Detection of copy-number variations: some DSDs are due to variations in the number of copies of certain genes (increased number of copies or deletions of alleles or chromosome loci). They are detected by methods such as multiple ligation-dependent probe amplification (MLPA) or microarray-based comparative genomic hybridisation (aCGH).11
- –
Lastly, whole exome sequencing (WES) or whole genome sequencing will be performed if there is no clear candidate gene, the results obtained with gene panels are normal, or in the context of studies seeking to identify novel candidate genes. These methods provide substantial amounts of information and must be used in the framework of strict technical and ethical protocols, with participation of the multidisciplinary team that will contribute to the interpretation of results.
Tables 2–4 summarise the different types of DSDs classified according to karyotype, phenotype, internal and external genitalia, biochemical abnormalities and the recommended approach to diagnosis.
ManagementGender assignmentGender assignment is a complex and critical decision in the management of DSDs. The decision rests with the parents, counselled by a multidisciplinary team (paediatric endocrinologists, surgeons, paediatric urologists, gynaecologists, neonatologists, geneticists, psychologists, social workers etc.). It is essential that this process be managed in reference centres with teams experienced in the management of DSDs, taking into account, among others, cultural and religious factors in different populations and the impact of the decision on the adult life of affected individuals (gender dysphoria, gonadectomy, dissatisfaction with the appearance of genitalia, etc.).
Historically, the approach consisted of early surgery with the aim of achieving cosmetically normal external genitalia and excising the gonads to match the assigned gender. However, in recent years elective surgery has been deferred to allow the patient to participate in decision-making in regard to both gender assignment and surgical intervention. In cases where it is possible to defer surgery for DSD and where families wish to take this approach, it is essential to establish a non-surgical care plan to help parents and patients cope with the social pressure associated with a child having atypical genitalia.
The current criteria for gender assignment are based on (1) psychosexual outcomes in adults with an aetiological diagnosis, (2) the potential for fertility, (3) surgical options and (4) the need for hormone replacement therapy during puberty.
Table 5 summarises the current recommendations for gender assignment12–24 applying the DSD classification of the 2006 Chicago Consensus Statement.1,2
Recommendations for gender assignment in individuals with a DSD.
| DSD | Proposed gender | Rationale |
|---|---|---|
| Sex chromosome DSD: | ||
| 45,X or Turner syndrome | Female | |
| 47,XXY | Male | |
| 45,X/46,XY or mixed gonadal dysgenesis | Female or male | |
| 46,XX/46,XY or ovotesticular DSD or chimerism | Female or male | |
| 46,XX DSD | ||
| CAH due to 21-hydroxylase deficiency | Female | Gender dysphoria is extremely rare when the female gender is assigned. The widespread approach is to assign the female gender at birth with early feminising surgery, although male gender assignment is considered in some cases with extreme virilisation.8,9 |
| 46,XX ovotesticular DSD | Female or male | These patients have functioning gonads and internal genitalia of both sexes. This disorder is more prevalent in Africa (51% of DSDs in África10), where due to cultural factors most of these individuals are assigned the male gender. However, countries outside Africa tend to assign the female sex. |
| 46,XX testicular DSD | Male | |
| 46,XY DSD | ||
| Complete androgen insensitivity syndrome (CAIS) | Female | 1. These patients have female psychosexual development. Gender dysphoria is very rare.11,12 2. They do not require surgery to correct the external genitalia. 3. Although replacement therapy may be required during puberty if gonadectomy is performed, testosterone replacement is ineffective in cases of complete androgen insensitivity. |
| Complete gonadal dysgenesis (Swyer syndrome) | Female | 1. These patients have female psychosexual development. 2. They can become pregnant with implantation of fertilised donor eggs and supportive hormone therapy. 3. High risk of malignant transformation of the gonads, and streak gonads should be resected, regardless of gender assignation. 4.- They do not require feminising genitoplasty. |
| 17-β-Hydroxysteroid dehydrogenase deficiency | Male | 1. They are highly likely to identify as male.13,14 2. They experience virilization during puberty (if gonads are present). 3. There are no reported cases of fertility. 4. Intermediate risk of germ cell tumour. |
| 5-α-Reductase | Male | 1. They are highly likely to identify as male.14 2. They experience virilization during puberty (if gonads are present), although there is little response from the micropenis. 3. Possible fertility. 4. Low malignancy risk. |
| Leydig cell hypoplasia (defect in LH receptor) | Female | 1. There are few data on psychosexual outcomes. 2. There are no published cases of fertility. 3. The risk of germ cell tumour is unknown. 4. If the female gender is assigned (complete forms), genitoplasty is not necessary as long as gonadectomy is performed before puberty. 5. If assigned the male gender, they require masculinising genitoplasty and androgen therapy to achieve a male appearance. |
| Partial forms (partial androgen insensitivity [PAIS], partial defects in androgen synthesis [5αR and 17β-HSD-3], partial dysfunction of the LH receptor and partial gonadal dysgenesis) | Male in cases of partial defect of 5αR and 17β-HSD-3. In all other cases, male or female19,21–24 | 1. 23% (9/54) are dissatisfied with the assigned gender, with a similar proportion of gender dysphoria in patients assigned to either gender15; except in cases of 5αR and 17β-HSD-3 deficiency, in which male gender assignment is recommended. 2. There are few long-term studies on gender dysphoria in these patients. 3. In most individuals, fertility is unlikely, regardless of the assigned gender. In individuals with PAIS, fertility is possible if the testes are not resected.16 Fertility is also possible in individuals with partial gonadal dysgenesis (PGD) with implantation of a fertilised donor egg if the uterus is sufficiently developed; it must be taken into account that there is an intermediate risk of developing germ cell tumours if the testes are preserved. |
Cases that present with adrenal insufficiency (glucocorticoids and/or mineralocorticoids) require immediate initiation of replacement therapy with administration of hydrocortisone.6 When it comes to sex steroid replacement for induction of puberty, there is no widespread agreement as to the time it should be initiated, the initial dose or the rate at which the dose should be increased. Most research groups emphasise the need to initiate pharmacologic treatment at low doses that are then progressive increased, but they disagree on the age at which it should start and the dosage in the first years of treatment. Generally, it is agreed that puberty should be induced when girls reach an approximate bone age of 11 years and boys a bone age of 12 years, increasing the dose at a slow pace.
The DSD Guideline4 presents the most widely approved approach to sex steroid replacement therapy during puberty in pp. 35 and 37 (Tables 6 and 7 of the guideline). It also provides detailed recommendations for treatment in regards to (1) testosterone treatment in prepubertal boys to promote penile growth; (2) Klinefelter syndrome; (3) Turner syndrome; (4) complete androgen insensitivity syndrome (CAIS); (5) partial androgen insensitivity syndrome (PAIS); (6) 5α-reductase deficiency; (7) 17β-hydroxysteroid dehydrogenase deficiency; (8) 46,XX gonadal dysgenesis; (9) 46,XY complete gonadal dysgenesis; (10) 46,XY partial gonadal dysgenesis, and (11) persistent Müllerian duct syndrome.
Surgical treatmentSurgical treatment of DSDs frequently involves irreversible changes to the patient's phenotype.1 The decision to choose this approach must be made jointly by the family and the multidisciplinary team advising the family. Where possible, the patient should be involved in the decision-making process. For this reason, in recent years there has been a growing tendency to defer surgery until the patient is of an age where they can become involved.25,26 In cases where surgery is performed at early ages, mutilating and irreversible procedures should be avoided.
Last of all, there is unanimous agreement that these procedures should only be performed by specialised surgeons in hospitals with considerable experience in the field.
The DSD Guideline4 specifies the different types of surgical intervention that can be performed for treatment of DSD by age and gender, including genital tubercle surgery (pp. 40 and 41), vaginoplasty (pp. 41 and 42), breast surgery (p. 48), excision of Müllerian duct remnants (p. 48), orchidopexy (p. 47) and gonadal cryopreservation.
The decision whether to perform prophylactic gonadectomy is complex. It must be made on a case-by-case basis taking into account its potential risks and benefits as well as: (1) the assigned gender, if different from gonadal sex; (2) the risk of malignant transformation, and (3) gonadal function (hormonal function and potential fertility).27
The DSD Guideline details the risk of malignant transformation based on the DSD classification (pp. 43–47).4
There is no widespread consensus when it comes to gonadectomy, although it is agreed that it should be considered in cases of gonadal dysgenesis or dysplasia, especially when the location of the gonads is intraabdominal.28
Transition to adult careThe need to guarantee a seamless transition from paediatric to adult care has been highlighted in several paediatric specialties, and patients with DSDs are particularly vulnerable at this juncture.29
There are published guidelines for the transition of patients with specific DSDs, such as Turner syndrome,30,31 Klinefelter syndrome32,33 or CAH.34,35
- •
Some of the general aspects of the transition to adult care in patients with DSDs are:
- •
Genital examination: in women with DSDs, a vaginal examination should be considered at some point during the followup. Women do not always require lengthening of the vagina, and should receive guidance in contemplating whether they wish to have sexual intercourse with penetration and when to start engaging in sexual relations.29
- •
Gonadectomy and risk of malignant transformation: in patients with DSDs at risk of malignant transformation of the gonads, the patient must be informed of this risk and be followed up in adult care.36
- •
Psychological problems: several studies have evinced the overwhelming need for psychological support of patients with DSDs during adolescence, most likely extending into adulthood.37,38
- •
Information about the diagnosis: patients will most likely receive detailed information about their condition when the time to transition to adult care nears.39 Information on the prognosis must include a discussion of potential fertility and the possibility of having offspring through the use of different assisted reproductive technologies. A better understanding of their condition will allow adolescents or young adults to seek support groups or additional support through social networks.
The proposed protocol for transition to adult care in patients with DSDs can be found in pp. 51 and 52 of the DSD Guideline.4
RegistersMultidisciplinary teams are unable to establish the aetiology of DSDs in 40% to 50% of cases, especially in patients with a 46,XY karyotype. Considering the diversity and complexity of DSDs and the need to investigate further unknown causes, health professionals need to share their knowledge, or, in other words, record information in registers.40
In Spain, there are two DSD registers that are currently active and accessible. One is a Spanish register, and the other an international one:
- •
The Spanish Register of DSDs-CAH is integrated in the Register of Rare Diseases of the Instituto de Salud Carlos III (ISCIII) in Madrid (https://registroraras.isciii.es).
- •
The International DSD Registry (I-DSD Registry, https://www.i-dsd.org/).
In Spain, there are two patient associations affiliated to the Federación Española de Enfermedades Raras (Spanish Federation of Rare Diseases, FEDER) that include individuals affected by some type of DSD:
- •
The Androgen Insensitivity Support Group (Grupo de Apoyo al Síndrome de Insensibilidad a los Andrógenos, GrApSIA), established in 2000 (https://grapsia.org/).
- •
The Asociación Española de Hiperplasia Suprarrenal Congénita (Spanish Association of Congenital Adrenal Hyperplasia), established in 2013 (http://hiperplasiasuprarrenalcongenita.org/).
- •
Lee et al. list other organisations at the international level.3
There are two working groups devoted to DSDs in the Sociedad Española de Endocrinología Pediátrica (Spanish Society of Paediatric Endocrinology, SEEP): one on CAH, and one on the rest of DSDs (the working group that authored this guideline). The Sociedad de Endocrinología y Nutrición (Spanish SEEN) has a working group devoted to gender identity (GIDSEEN) that also investigates DSDs, although its main focus is transsexuality.
In Spain, the Ministry of Health, Social Services and Equality is considering the need to establish a system for the accreditation of reference centres, departments and units devoted to endocrinological diseases. The two Spanish endocrinological societies (SEEP and SEEN) and the Asociación Española de Pediatría (Spanish Association of Paediatrics, AEP) have presented an initial set of files corresponding to several diseases, including one for DSDs (2017).
Since 2017, European Reference Networks for Rare Diseases (ERN) have been established in Europe. One is the Rare Endocrine Diseases Reference Network (EndoERN), which has a main thematic group named Sex Development and Maturation that is dedicated to DSDs (http://endo-ern.eu/).
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Guerrero-Fernández J, Azcona San Julián C, Barreiro Conde J, Bermúdez de la Vega JA, Carcavilla Urquí A, Castaño González LA, et al. Guía de actuación en las anomalías de la diferenciación sexual (ADS)/desarrollo sexual diferente (DSD). An Pediatr (Barc). 2018;89:315.
