








Noonan syndrome (NS) is a relatively common genetic condition characterised by short stature, congenital heart defects, and distinctive facial features.
NS and other clinically overlapping conditions such as NS with multiple lentigines (formerly called “LEOPARD” syndrome), cardiofaciocutaneous syndrome, or Costello syndrome, are caused by mutations in genes encoding proteins of the RAS-MAPKinases pathway. Because of this shared mechanism, these conditions have been collectively termed “RASopathies”. Despite the recent advances in molecular genetics, nearly 20% of patients still lack a genetic cause, and diagnosis is still made mainly on clinical grounds.
NS is a clinically and genetically heterogeneous condition, with variable expressivity and a changing phenotype with age, and affects multiple organs and systems. Therefore, it is essential that physicians involved in the care of these patients are familiarised with their manifestations and the management recommendations, including management of growth and development. Data on growth hormone treatment efficacy are sparse, and show a modest response in height gains, similar to that observed in Turner syndrome.
The role of RAS/MAPK hyper-activation in the pathophysiology of this group of disorders offers a unique opportunity for the development of targeted approaches.
El síndrome de Noonan (SN) es una enfermedad de origen genético relativamente frecuente cuyas manifestaciones fundamentales son la talla baja, la cardiopatía congénita y un fenotipo facial característico.
La causa del síndrome de Noonan y de otras enfermedades clínicamente solapadas como el síndrome de Noonan con lentiginosis múltiple (anteriormente llamado síndrome LEOPARD), el cardiofaciocutáneo o el síndrome de Costello, son mutaciones en genes que codifican para proteínas de la vía de señalización de las RAS-MAPKinasas. Debido a este sustrato común este grupo de enfermedades son denominadas colectivamente “rasopatías”. A pesar de los avances genéticos de las últimas décadas cerca de un 20% de pacientes no tienen causa genética identificada, y el diagnóstico sigue siendo clínico.
El síndrome de Noonan se caracteriza por una alta heterogeneidad clínica y genética, con afectación variable, y cambiante con la edad, de múltiples órganos y sistemas. Debido a esta variabilidad es fundamental que los médicos involucrados en su cuidado estén familiarizados con sus manifestaciones y conozcan las recomendaciones de seguimiento, incluido el seguimiento del crecimiento y desarrollo. Hasta la fecha los escasos datos de crecimiento con GH a talla adulta dan resultados de ganancia de talla moderados, semejantes a los obtenidos en síndrome de Turner.
La hiperactivación de la vía RAS-MAPK como base común de esta familia de enfermedades brinda una oportunidad única para el desarrollo de tratamientos dirigidos a la etiología de estos trastornos.
Noonan syndrome (NS) is an autosomal-dominant genetic disorder characterized by a distinctive phenotypic triad: craniofacial dysmorphic features resulting in a distinctive facial phenotype, congenital heart disease and short stature.1 First described by Dr Jacqueline Noonan more than 50 years ago, it is a relatively frequent disorder with an estimated incidence of 1 in 1000 to 2500 live births. Since 2001, it is known that mutations in genes that encode proteins involved in the intracellular RAS-MAPK (Mitogen Activated Protein Kinases) signalling pathway are responsible for NS and other genetic disorders with similar phenotypes, such as Costello syndrome, cardiofaciocutaneous syndrome, Noonan syndrome with multiple lentigines (NS-ML, formerly known as LEOPARD syndrome) or la neurofibromatosis type 1. Due to the overlap in phenotypic characteristics and the underlying molecular mechanisms of these diseases, they have been grouped under the label of “neurocardiofaciocutaneous syndromes” or, more generally, “RASopathies”.
Our aim was to synthesise the current evidence on this disease and propose guidelines for the follow-up of these patients from infancy to adulthood.
Clinical descriptionNoonan syndrome is characterised by multisystemic involvement, a broad phenotypic heterogeneity and a variable clinical presentation.
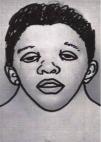
Craniofacial featuresIt is characterised by a distinctive facies that changes with age, with more striking features in children that become less prominent in adulthood.2 The characteristics features are a triangular face shape (broadened forehead with narrowing down toward the chin), low-set posteriorly rotated ears with a thick helix (90%), downslanting palpebral fissures (95%), hypertelorism, ptosis, strongly arched diamond shaped brows, usually blue or blue-green irises, nose with a flattened bridge and bulbous tip, a long and deep philtrum, wide peaks in upper lip (Cupid’s bow appearance), short neck, and low posterior hairline shaped like a W (55%) (Fig. 1). Infants may exhibit webbing of the neck.
Cardiovascular manifestations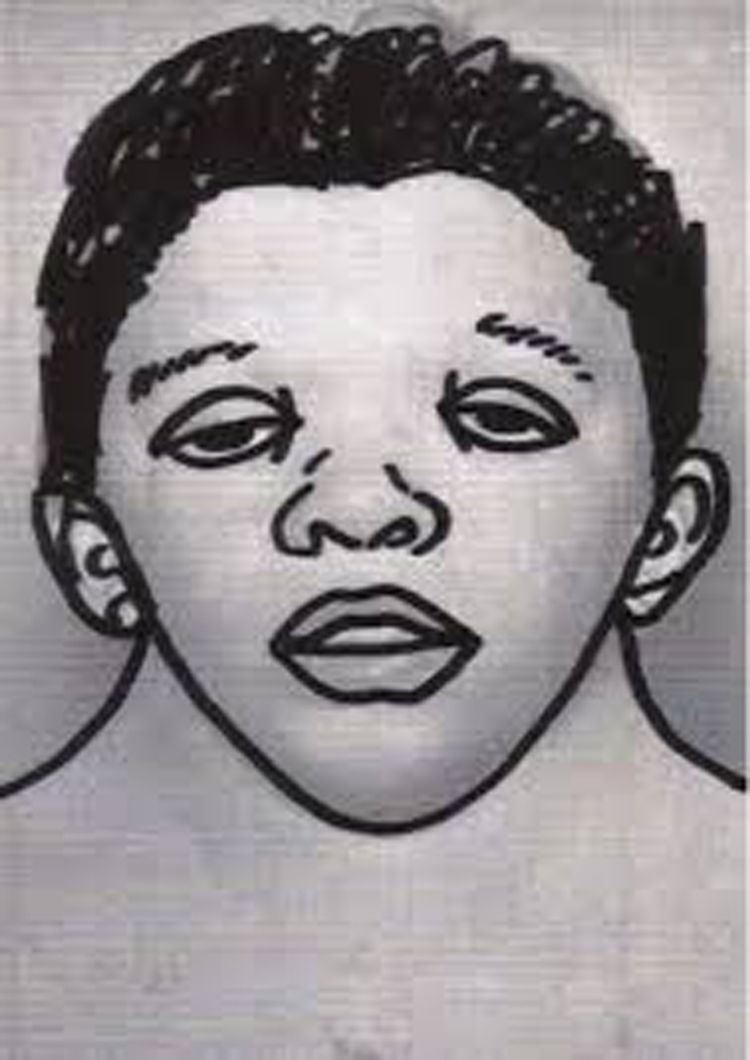
Cardiac involvement is one of the main clinical features of NS. The prevalence of cardiovascular abnormalities is estimated at 70%–80% of cases,1,3 although prenatal diagnosis is rare. A wide spectrum of abnormalities has been described, the most frequent being pulmonary stenosis, commonly associated with valve dysplasia (50%–60%), hypertrophic cardiomyopathy (20%)4, usually detected early in life,5 atrial septal defects (6%–10%) and ventricular septal defects, pulmonary artery branch stenosis, anomalies in the mitral valve or coronary arteries, coarctation of the aorta and, less frequently, tetralogy of Fallot and patent ductus arteriosus.
The literature also describes electrocardiographic abnormalities in these patients (50%).6 Frequently, these involve wide QRS intervals with a predominantly negative pattern in the left precordial leads and left axis deviation with giant Q waves, even in the absence of structural cardiac abnormalities. Arrythmias are infrequent.7
Impact on growth and developmentPrenatal growth is not usually affected, although there is a higher prevalence of small for gestational age compared to the general population.8
Children with NS exhibit a pattern of postnatal growth retardation characterised by a prepubertal growth in the 3rd percentile, delayed onset of puberty with attenuated growth spurts and an adult height approximately 2 standard deviations (SD) below the mean. Delayed puberty is common, with a mean age at onset of 13.4 years (10.8–16.4 years) in boys and 13 years (10.9–15 years) in girls. There is a mean delay in bone age of 2 years. Specific growth charts for children with NS have been developed,9 but none of them have been developed in the Spanish population.
Some authors have found hormone profiles suggestive of growth hormone deficiency in patients with NS, with decreased levels of insulin‐like growth factor 1 (IGF1) and insulin-like growth factor binding protein 3 (IGFBP-3) and responses to stimulation with growth hormone in the upper limit.10 Other studies suggest that hyperactivation of RAS-MAPK could affect chondrocyte differentiation during bone growth through an IGF1-independent mechanism.11
Nutritional aspects and disturbances of energy homeostasisIn the neonatal period, infants with NS exhibit difficulty feeding with weak sucking, prolonged duration of feedings and sporadic episodes of nausea and vomiting, in some cases requiring nasogastric tube feeding (25%), and gastroesophageal reflux. These symptoms usually improve or resolve completely by age 15 months.12,13
Several studies have described a tendency toward a low body weight not only in the first years of life, but through the lifespan. An animal model with the genetic variant p.Thr468Met (characteristic of NS-ML) with a metabolic phenotype characterised by defective adipogenesis, enhanced energy expenditure and mitochondrial activity, enhanced insulin sensitivity and resistance to the effects of an obesogenic diet.14 The implications of these findings are unclear, although future research will likely uncover further information on these disturbances in energy homeostasis.
Genitourinary and renal manifestationsCryptorchidism has been described in 60%–80% of male patients with NS.12 Several studies have reported elevated levels of luteinising hormone (LH) and follicle-stimulating hormone (FSH) and decreased levels of inhibin B and anti-Müllerian hormone (AMH) in patients with NS compared to the general population, with a high prevalence of a hormone pattern suggestive of Sertoli cell dysfunction both in patients with cryptorchidism and patient without it.15 This hypothesis is further supported by the critical role played by Src-homology 2 domain-containing tyrosine phosphatase 2 (SHP2) in maintaining Sertoli cell function. The literature also describes a higher frequency of infertility in male patients with NS.
In contrast, it appears that women with NS have normal fertility (which would explain why maternal transmission predominates in familial cases).
Renal abnormalities are present in 10% of cases (duplex collecting system, solitary kidney, pyeloureteral stenosis and renal pelvis dilation).16
Hypothyroidism and autoimmune disordersMost studies have found a higher incidence of autoimmune thyroiditis compared to the general population,13 although with levels similar to or slightly above subclinical hypothyroidism.17 Other features described in the literature include coeliac disease, systemic lupus erythematosus, vitiligo and anterior uveitis.
Musculoskeletal abnormalitiesChest deformities (with pectus excavatum inferiorly and pectus carinatum superiorly) occur in 70%–95% of cases, possibly accompanied by wide-spaced nipples, cubitus valgus, genu valgum,18 scoliosis (10%–15%) or other less frequent deformities of the spine, such as kyphosis, spina bifida or malformations of vertebrae or ribs. There is redundant skin in the neck and a prominent trapezius. Joint hypermobility is also common.13
There is evidence that the overall bone mineral density in children with NS is lower compared to controls matched for age, sex, height and ethnicity. However, the incidence of childhood fractures does not appear to be higher compared to the general population. The incidence of fractures in adults with NS is unknown, and therefore the clinical relevance of these findings is unclear and needs to be established through performance of longitudinal studies. Histomorphometric analysis of bone tissue from NS patients and mouse models of NS may further elucidate the relationship between the RAS-MAPK pathway and skeletal homeostasis.19
Blood disorders and cancerA tendency to develop bruises and bleeding is frequent, especially in childhood, although severe haemorrhage is rare (3%). Some of the haemostatic abnormalities in NS described in the literature are bleeding diathesis, factors VIII, XI and XII deficiency, low platelet count and platelet function defects, in isolation or combined.20 In many cases, there is no association between the results of coagulation tests and bleeding diathesis.
As is the case of all RASopathies, NS carries an increased risk of blood cancers and solid tumours that is actually estimated to be 8.1 times greater (95% confidence interval [CI], 3.5–16) compared to the general population.21 The most frequent malignancies are gliomas (dysembryoplastic neuroepithelial tumours), acute lymphoblastic leukaemia, neuroblastoma and rhabdomyosarcoma. Substitutions in codon 61 and substitution 218C > T (p.Thr73Ile) in the PTPN11 gene, the p.Thr58Ile variant of the KRAS gene and mutations in the CBL gene are associated with a higher incidence of juvenile myelomonocytic leukaemia (JMML), a severe myeloproliferative disorder.22 Cases of JMML tend to have an early onset and a benign course, and there are reports of spontaneous resolution in the literature. However, a recent study documented a mortality greater than expected in NS attributable to severe JMML in the neonatal period,23 which suggests that JMML may go undetected in this age group due to the fatal outcome of these cases.
Neurologic manifestations, cognitive and behavioural changesCognitive functioning is within the range of normality in up to 80% of cases, 24 with an intelligence quotient ranging from 70 to 120.2,25 There have been reports of deficiencies in the ability to identify and verbally express emotions (alexithymia). Other psychiatric disorders described in these patients include mood disorders, difficulties with communication and interpersonal interaction and attention-deficit hyperactivity disorder.13 Language impairments are more frequent in children with NS compared to the general population and, when present, are associated with an increased risk of reading and spelling difficulties.24
Orodental featuresFrequently described features include a high-arched palate (55%–100%), dental malocclusion (50%–67%), temporo-mandibular joint problems (72%) and micrognathia. There have been reports of class II malocclusion and an increased incidence of open bite and posterior crossbite compared to the general population. There may also be multiple unerupted permanent teeth, submerged deciduous teeth and supernumerary teeth.26 Multiple giant cell lesions are infrequent, but highly suggestive of NS.
Lymphatic disordersLymphoedema occurs in fewer than 20% of cases, but when present causes significant morbidity. Peripheral lymphoedema may be present as early as birth (palms and soles) and resolve in the early years of life.27 Other, less frequent lymphatic abnormalities may include hydrops fetalis, pulmonary, intestinal or testicular lymphangiectasia, chylothorax or chylous ascites, hypoplastic inguinal and iliac lymphatic vessels, absent or atretic thoracic duct or localised lymphoedema in the scrotum or vulva.16,28
Cutaneous manifestationsPatients with NS may present with pigmented nevi (25%), café au lait spots (10%) and lentigines. Keratosis pilaris is common (in the upper arms), and in cases in which it affects the face, the eyebrows may be absent.29 The skin is hyperelastic, the hair thick and curly and the nails dystrophic. Foetal pads on fingers and toes are common (67%).13
Vision and hearing problemsOcular abnormalities are frequent (95%): strabismus (40%–65%), refractive errors (60%), amblyopia (33%) or nystagmus (10%). Anterior segments may occur (60%), including cataracts. Other possible ocular abnormalities are optic nerve head or disc drusen (20%), optic nerve hypoplasia and coloboma.13,30 Hearing loss secondary to otitis media is a frequent complication (15%–40%). Sensorineural hearing loss is less frequent.31 There have been descriptions of vestibular anomalies and structural anomalies of the inner ear in some cases.
Prognosis and quality of lifeLong-term data are scarce. There is evidence that feeding difficulties in infancy could be an early predictor of future language delay. One study found that one third of adults with NS had attended a school for children with learning disabilities and 20% a conventional school while receiving academic support. Forty-three percent of participants in the study had achieved an educational certification (lower level secondary general education certificate or higher). The reported data on health-related quality of life has shown no difference relative to the general population.32
Adults with NS require long-term cardiological follow-up. An absence of symptoms, a normal cardiac output and pulmonary arterial blood pressure and a right ventricular pressure of less than 100 mmHg are associated with a good prognosis.33 The mortality associated to hypertrophic cardiomyopathy is the same in patients with NS and patients without NS. The mortality was 9% at a mean age of 60 years.15
Genetics of Noonan syndrome and RASopathiesThe RAS-MAPK pathwayThe RAS-MAPK cascade is a well-known intracellular signalling pathway triggered by the binding of extracellular ligands such as hormones, cytokines and growth factors end resulting in transcription in the cell nucleus, and is involved in cell proliferation, differentiation and apoptosis processes. RAS works as a molecular switch that stimulates the sequential activation of kinases in the MAPK pathway (RAF, MEK and ERK) that eventually results in changes in gene transcription (Fig. 2). Most of the mutations described in these genes lead to a gain of function, and at present it is believed that RASopathies result from such gain of function or at least dysregulation of the RAS-MAPK pathway. The pattern of inheritance of NS is autosomal dominant, although recently there have been reports of biallelic variants of LZTR1 associated with autosomal recessive NS.
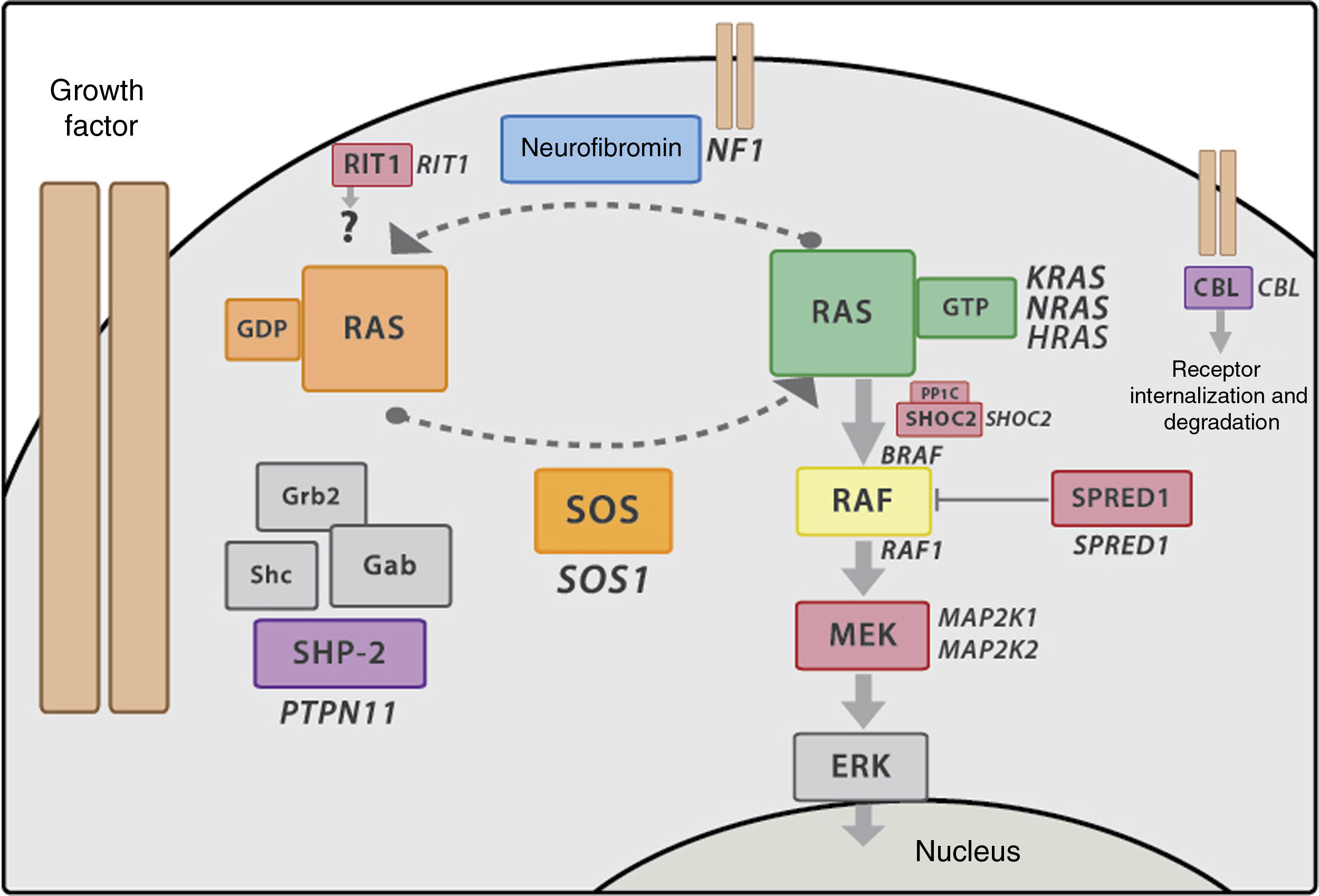
RAS-MAPK signalling cascade. After the ligand binds the cell-surface receptors, the intracellular portion of the receptor is phosphorylated and recruits adaptor proteins such as GRB2, which form complexes with guanosine exchange factors (such as SOS) that promote conformational change of the inactive protein RAS bound to GDP to the active form bound to GTP. The active RAS-GTP form then activates different isoforms of RAF (RAF1, BRAF), MEK (MEK1, MEK2) and lastly, ERK.
To date, mutations in more than 20 genes have been described in association with NS, the RASopathy with the greatest known locus heterogeneity. Yet, the RASopathies are monogenic disorders and only rare cases have been reported where patients had mutations in more than 1 gene. Table 1 presents the genes currently known to be involved in NS. Given the growing number of identified genes and how many laboratories have the capability for massive gene sequencing, efforts must be made to confirm the role of identified genes and each of the variants identified for each gene. The RASopathy Expert Panel of the Clinical Genome Resource (ClinGen), funded by the National Institutes of Health of the United States, has recently published a guideline for the interpretation of 19 genes involved in RASopathies34 that also provides a classification of the strength of the clinical and experimental evidence available on the different variants and the correlated phenotypes.
List of syndromes associated with mutations in genes in the RAS-MAPK pathway.
| Syndrome | Features | Gene | % | ClinGen | Locus |
|---|---|---|---|---|---|
| NoonanOMIM #1639501:1000/2500 | Craniofacial abnormalitiesCongenital heart disease (70%–80%):Pulmonary valve stenosis 50%–60%Hypertrophic cardiomyopathy 20%–25%Septal defects 10%–20%Short stature (70%, usually mild)Oncological and haematologic disordersEasy bleeding.Transient myeloproliferative disorder in newbornsJuvenile myelomonocytic leukaemiaIntellectual disability (10%–30%)Usually mildCNS abnormalitiesSeizures (5%–15%), Chiari malformation I (infrequent)Musculoskeletal anomalies Chest deformities, joint hypermobility, scoliosis, hypotonia,Vision and hearing problemsRefraction errors, strabismus, hearing loss (infrequent)Genitourinary disordersCryptorchidism.Sertoli cell dysfunction and infertility in males. | PTPN*11 | 50 | Definitive | 12q24.1 |
| SO*S1 | 11 | Definitive | 2p22.1 | ||
| RAF*1 | 5 | Definitive | 3p25.1 | ||
| BRA*F | < 2 | Moderate | 7q34 | ||
| MAP*2K1 | < 2 | Limited | 15q22.31 | ||
| KRA*S | 1,5 | Definitive | 12p12.1 | ||
| NRA*S | 0,2 | Definitive | 1p15.2 | ||
| RIT*1 | 5 | Definitive | 1q22 | ||
| SHO*C2 | 2 | Disputed | 10q25 | ||
| PPP*1CB | No evidence | 2p23 | |||
| SO*S2 | Moderate | 14q21.3 | |||
| RR*AS | Limited | 19q13.33 | |||
| RAS*A2 | Limited | 3q23 | |||
| SP*RY1 | No evidence | 4q28.1 | |||
| LZT*R1 | Strong (limited for AR) | 22q11.21 | |||
| MA*P3K8 | No evidence | 10p11.23 | |||
| MY*ST4 | No evidence | 10q22.2 | |||
| A2*ML1 | Disputed | 12p13.21 | |||
| RA*SA1 | Disputed | 5q14.3 | |||
| M*RAS | Limited | 3q22.3 | |||
| CB*L | No evidence | 11Q23.3 | |||
| Noonan syndrome with multiple lentigines (formerly LEOPARD)OMIM #1511001:100000 | Hypertrophic cardiomyopathy 60%–70%Short stature < 50%, usually mildChildhood cancer < 1%Multiple lentigines, café au lait spotsSensorineural hearing loss 20% | PTP*N11 | 90 | Definitive | 12q24.1 |
| RA*F1 | < 5 | Limited | 3p25.1 | ||
| BR*AF | < 1 | Limited | 7q34 | ||
| Cardiofaciocutaneous syndromeOMIM #1151501:200000 | Bitemporal narrowing, facial coarsenessHigher prevalence of endodermal anomalies (palmoplantar hyperkeratosis, multiple melanocytic nevi)Mild to severe intellectual disability in > 95%Crisis 50%, hydrocephalus/ cortical atrophy, neuropathyJoint contractures, scoliosis (30%–40%)Optic nerve anomaliesSevere feeding difficulties | BR*AF | 75 | Definitive | 7q34 |
| KR*AS | < 5 | Strong | 12p12.1 | ||
| MAP*2K1 | 25 | Definitive | 15q22.31 | ||
| MA*P2K2 | Definitive | 19p13.3 | |||
| Costello syndromeOMIM #2180401:400000 | Hypertrophic cardiomyopathy 60%–70%. Multifocal atrial tachycardiaFacial coarsenessModerate short stature in >95%Solid tumours (15%–20%)Soft and loose skin, deep creases in palms and soles of feet, multiple papillomas and verrucous lesionsSeizures in 20%–50%, Chiari malformation I, syringomyelia, hydrocephalusUlnar deviationSevere feeding difficulties | H*RAS | >95 | Definitive | 11p15.5 |
| Noonan syndrome-like disorder with loose anagen hairOMIM #607721About 100 reported cases | Loose anagen hairMitral valve dysplasia, septal defectsHyperactivityGH deficiency | SH*OC2 | >95 | Definitive | 10q25 |
| PP*P1CB | Strong | 2p23 | |||
| Neurofibromatosis type 1 (NF1)OMIM#1622001:3000 | Clinical criteria for diagnosis of NF1 | NF1 | >95 | No evidence | 17q11.2 |
| Legius syndromeOMIM#611431About 200 reported cases | Cutaneous criteria for diagnosis of NF1 in absence of neurofibromas | SP*RED1 | >95 | No evidence | 15q14 |
ClinGen: Assessment of the gene-disease association of 19 genes with different RASopathies using the by the RASopathy Expert Panel of the Clinical Genome Resource34 using the semiquantitative classification of the available genetic and experimental evidence published by Strande et al: Definitive (12−18 and upheld over time); strong (12−18); moderate (7–11); limited (1–6); no reported evidence (0); disputed (conflicting evidence from additional cases after the original publication raise about the role of the gene in the aetiology of the disease, without refuting it); refuted. No evidence: genes not included in the evaluation of the Clinical Genome Resource. AR, autosomal recessive inheritance pattern: very rare cases of Noonan syndrome in patients with biallelic mutations in the LZTR1 gene that seem to exhibit this inheritance pattern have been described.
There are multiple descriptions of genotype-phenotype correlations in NS in the literature, although there are no exclusive phenotypic features of any given genotype, probably due to genetic and epigenetic factors that influence penetrance and expressivity. Table 2 presents a brief description some of the most widely accepted genotype-phenotype correlations.
Genotype-phenotype correlation.
| Genotype-phenotype correlation | |
|---|---|
| Gene | Associated clinical features |
| PTPN11 | Typical facies. Pulmonary valve stenosis. Easy bruising. Cryptorchidism. Family history of NS |
| SOS1 | Cutaneous features characteristic of cardiofaciocutaneous syndrome (keratosis pilaris, thin and/or curly hair, sparse brows)Low prevalence of short stature and intellectual disability |
| RAF1 | Hypertrophic cardiomyopathy (neonatal in some cases). Pigmented nevi |
| KRAS | Cognitive impairment, cutaneous features characteristic of cardiofaciocutaneous syndrome |
| BRAF | Cognitive impairment, cutaneous features characteristic of cardiofaciocutaneous syndrome |
| CBL | Increased prevalence of JMML and solid tumoursLow prevalence of short stature, congenital heart defects or cryptorchidism |
| RIT1 | Hypertrophic cardiomyopathy (neonatal in some cases). JMMLLow prevalence of short stature, cutaneous manifestations and intellectual disability |
| Variant-phenotype correlation | |
|---|---|
| Variant (gen) | Associated features* |
| p.Ser2Gly (SHOC2) | Noonan syndrome with loose anagen hair: hair that comes off easily by pulling during the growth or anagen phase, increased frequency of septal and mitral valve defects, ectodermal anomalies, hyperactivity, nasal voice, growth hormone deficiency |
| p.Thr468Met (PTPN11) | Noonan syndrome with multiple lentigines: lentigines, hypertrophic cardiomyopathy, hearing loss, preserved stature |
| p.Thr73Ile, substitutions at p.Asp61 (PTPN11) | Increased risk of JMML |
While the notion of a strong correlation between a given gene and a specific phenotype is appealing from a theoretical perspective, clinical experience suggests that reality is far more complex. For this reason, the classification of RASopathies based on clinical features currently continues to be favoured over a classification based on the involved genes.
Diagnosis and differential diagnosisDespite advances in molecular testing, NS is diagnosed essentially based on clinical features, and an underlying genetic mutation is not identified in 20%–30% of patients. The variability in its phenotypic expression, the overlap with other diseases and the changes in phenotype that take place with age pose a significant challenge in its diagnosis, especially in the first year of life. The diagnostic criteria that are now universally accepted were first developed by van der Burgt in 1994 and revised in 2007 (Table 3).29
Criteria for diagnosis of Noonan syndrome.
| Feature | A = Major criteria | B = Minor criteria | |
|---|---|---|---|
| 1 | Facial | Typical face dysmorphology (varies with age) | Suggestive face dysmorphology |
| 2 | Cardiac | Pulmonary valve stenosis, hypertrophic obstructive cardiomyopathy and/or ECG typical of NS | Other defect |
| 3 | Height* | < 3rd percentile | < Percentil p10 |
| 4 | Chest wall | Pectus carinatum/excavatum | Broad thorax |
| 5 | Family history | First degree relative with definite NS | First degree relative with suggestive NS |
| 6 | Other | All of the following: Mental retardation, cryptorchidism and lymphatic dysplasia | One of the following: Mental retardation, cryptorchidism and lymphatic dysplasia |
Noonan syndrome:
A (typical face dysmorphology) plus one other major criteria (2A-6A) or 2 minor criteria (2B-6B);
B (suggestive face dysmorphology) plus 2 major criteria (2A-6A) or 3 minor criteria (2B-6B).
The differential diagnosis must include other RASopathies as well as other syndromes unrelated to the RAS-MAPK pathway, such as Aarskog syndrome, Turner syndrome, Baraitser-Winter syndrome and actinopathies. While other RASopathies must be considered in the differential diagnosis, they are much less prevalent than NS with the exception of neurofibromatosis type 1.
Recommendations for follow-upIt is important that the different specialists potentially involved in the management of these patients be knowledgeable of the clinical features and potential complications associated with NS. Having a physician in charge of coordinating the care delivered by different specialists is helpful. This role could be assigned to the clinical geneticist or otherwise any other physician (primary care paediatrician, endocrinologist or cardiologist) that is particularly involved in the management of the patient, and the aim is to ensure that the multiple aspects that require care and follow-up are being covered. Table 49,28,29 offers a summary of the recommendations for management of patients with NS by age group.
Recommendations for follow-up.
| Birth-1 year | 1−5 years | 6−11 years | 12−18 years | Adulthood | |
|---|---|---|---|---|---|
| Diagnosis | |||||
| Evaluation of phenotype | Discuss findings with parents* | ||||
| Molecular testinga | *r | ||||
| Genetic counselling | Parents | Patient | Patient | ||
| Growth | |||||
| Feeding and weight gain | Every 1−3 months b | b,c | |||
| Monitor growth in general and specific growth charts d | Every 1−3 months | Every 6−12 months | Every 6−12 months e | Every 6−12 monthse | |
| Cardiological evaluation | * + s,t | * + s,t | * + s,t | * + s,t | *stf |
| Hearing assessment g | At age 6−12 months t | Yearly | Yearly | ||
| Ophthalmological assessment | * + s t | s | + s t | s | t |
| Neurologic evaluationh | Yearly i | Yearly | Yearly | Yearly | |
| Scoliosis monitoring j | Yearly | Yearly | Yearly | ||
| Abdominal ultrasound k,l | * | ||||
| Testicular descent | From 6 months | Yearly m | Yearly | Yearly | n |
| Coagulation testso | In case of surgery | In case of surgery + | In case of surgery + | In case of surgery* | In case of surgery* |
| Thyroid function assessment | Every 3−5 years | Every 3−5 years | Every 3−5 years | ||
| Psychomotor development evaluationEmphasis | Every 1−3 monthss tHypotonia and motor skills | Every 6−12 months,Spoken language s | YearlysAcademic performance p | Yearly sAcademic performance16 | |
| Skin problems q | + s | + s | + s | + s | + s |
| Dental evaluation | s t | s t | s t | s t | |
| Social support/refer to support groups | * + | * + | * + | * + | *+ |
We recommend that a clinical geneticist guide the diagnostic process and the initial genetic evaluation. The expected proportion of positive results in patients with a definitive clinical diagnosis of NS is 70%–80% if all genes with a known association are sequenced. If a pathogenic variant is identified, testing of parents is recommended to make an accurate prediction about the risk of transmission to the offspring.
Refer to specialists for evaluation of feeding and swallowing if necessary. Refer to speech therapist if necessary. Repeated vomiting should be evaluated to rule out gastroesophageal reflux and intestinal malrotation. Medical treatment for reflux should be given as needed. Persistent vomiting or food refusal may require enteral feedings through a nasogastric tube or gastrostomy.
Use specific growth charts for Noonan syndrome with caution, as they are not genotype-specific. Rule out growth hormone deficiency, as would be done in the general population. Treatment with growth hormone should be individualised. In case of initiation of growth hormone therapy, perform a cardiological and haematological assessment at baseline and monitor the patient closely thereafter.
The possibility of delayed onset of puberty should be anticipated, offering support and counselling. In case of a significant delay (absence of breast development in girls at age 13 years or testicular volume < 4 mL measured with a Prader orchidometer in boys aged more than 14 years), perform an evaluation of gonadal function. Due to the association with Sertoli cell dysfunction, testicular volume may not provide an accurate reflection of the stage of pubertal development.
Evaluation including an echocardiogram even if previous evaluations have been normal. We recommend an evaluation every 5 years in patients without a previous history of cardiac disease. In patients with congenital heart disease, the follow-up should adhere to the recommendations of the cardiologist. Long-term follow-up is essential in patients that undergo cardiac surgery or a valvuloplasty.
Refer for a hearing assessment between 6 and 12 months of age. Monitor patients closely and treat episodes of otitis aggressively to prevent hearing loss.
Maintain a high level of suspicion in the evaluation of neurologic features (consider the possibility of Chiari malformation or hydrocephalus if the patient has headache or other neurologic symptoms and schedule an MRI if either are suspected). Epilepsy should be management per the protocol applied to the general population.
Consider referral to paediatric neurologist for formal neurodevelopmental evaluation and to early intervention programme including occupational and physical therapy. It is recommended that parents be informed that in patients with NS, motor delay is frequently due to hypotonia and is not always associated with learning disorders or intellectual disability in adulthood.
Determine the presence or absence of hepatosplenomegaly. In case of splenomegaly, perform a complete blood count. In case of hepatosplenomegaly, perform a complete blood count and liver function panel. In patients with variants that carry a high risk of cancer (en p.61 substitutions and the p.Thr73Ile mutation in the PTPN11 gene, and the p.Thr58Ile KRAS variant) consider clinical evaluations with performance of a complete blood count every 3 months in the first year of life.
Annual evaluation to check for potential testicular retraction in patients with previously descended testicles. Testicular maldescent is managed following the protocol applied to the general population.
Inform of the increased risk of infertility in men with NS, including those without a history of cryptorchidism. Refer to fertility clinic or endocrinologist as needed.
In patients with Noonan syndrome, the correlation between the results of coagulation tests and the tendency to bleed easily is weak, so extreme caution should be exerted if in case of surgical intervention, suspending treatment with acetylsalicylic acid prior to surgery if the patient is receiving it, and considering consultation with a paediatric haematology specialist to assess the risk of haemorrhage and determine the measures to be taken to reduce this risk.
In patients with a positive genetic test, we recommend that clinicians periodically obtain updated information on the clinical manifestations associated with the specific variant, especially in case of rare mutations, consulting with an expert clinical geneticist as needed.
Lastly, in patients with RASopathies other than NS or patients with NS associated with uncommon mutations, consider follow-up in multidisciplinary units with experience in these diseases.
Growth hormone replacement therapyOne of the options for symptomatic treatment of NS is recombinant human growth hormone (rhGH). Its use for this indication was approved by the United States Food and Drug Administration in 2007, and more recently by the European Medicines Agency (https://mri.cts-mrp.eu/Human/Product/Details/14639), and the Spanish Agency for Medicines and Medical Devices (Agencia Española de Medicamentos y Productos Sanitarios, AEMPS) (https://cima.aemps.es/cima/dochtml/ft/62977/FT_62977.html). However, we ought to highlight that no randomised clinical trials with follow-up through achievement of final height have been published to date.
T studies in the literature are difficult to compare due to the heterogeneity of the methodologies used, patient ages, dosage and duration of treatment. Overall, they have reported a significant increase in final height z-score of 1.4 ± 0.8 (corresponding to 9.5 ± 5.4 cm),35–38 although some studies reported significantly more modest increases (Table 5). Most studies have found better outcomes the longer the duration of treatment in the prepubertal period. Consistently with the hypothesis that growth impairment in NS is due to growth hormone resistance, some studies have reported poor responses in patients with a mutation in the PTPN11 gene,35 although other studies have not corroborated this result.37 While growth hormone deficiency has been described in some patients with NS, this does not seem to be the main reason for growth impairment in these patients, with the exception of those with a SHOC2 mutation, who frequently have this deficiency. When it comes to dosage, recent studies have found an improved short-term response at higher doses,37 although there are no data on long-term safety and efficacy. In short, the following are reasonable recommendations based on the current medical literature:
- none-
Independently of the conditions established for the indication for treatment once it is approved, the decision to use rhGH in patients with NS should be made on a case-by-case basis. Factors to be taken into account include the height, age (early initiation is preferred to maximise prepubertal linear growth), the presence of comorbidities (hypertrophic cardiomyopathy is not a contraindication but would require rigorous cardiological follow-up; scoliosis would require close monitoring as it could worsen during treatment; nutrition should be assessed and energy intake deficits resolved before initiation of treatment, and in case of clinical features compatible with growth hormone deficiency, evaluation of the somatotropic axis should be considered) and the genotype (in patients with variants associated with a high risk of cancer, the use of rhGH should be considered with utmost caution and, if initiated, requires close monitoring).
- none-
The recommended initial dose is 33 μg/kg/day (to be increased to up to 66 μg/kg/day in case of poor response), and especial attention should be paid to IGF1 levels, carbohydrate metabolism and other possible adverse events. If the patient exhibits a poor response despite 1–2 years of treatment at high doses, consider discontinuation of treatment, as the peak response should occur in the early years of treatment. Patients with a clinical diagnosis with negative genetic test results should be considered eligible for treatment, although in these cases assessment by a clinical geneticist with experience in the RASopathies is recommended, as is the maintenance of a watchful and critical attitude throughout treatment.
Studies assessing the use of growth hormone in Noonan syndrome through the final height (adapted from Carcavilla 2014).
| Reference | Design | Genetic test | Duration (years) | Age at initiation (years) | Height z at baseline | Dose(μg/kg/day) | ΔHeight z 1 y | Age/definition of final height | Final height | Δ final height |
|---|---|---|---|---|---|---|---|---|---|---|
| Kirk 2001 | Observational KIGS | No | 5.3 | 10.2 ± 3.3 | −2.9 ± 0.7 | 40 ± 20 | 0.4 | ♂ > 17 years♀ > 15 yearsGV< 2.5 cm/year | (n = 10)♂159.9 cm(z = -2.2)♀147.2 cm(z = -2.5) | 3.1 cm(0.8 SDs) |
| Osio 2005 | Uncontrolled clinical trial | No | 7.5 | 7.78.6 | −3.1 ± 0.5−2.7 ± 0.4 | 3366 | 0.8 ± 0.4 | 19.5 ± 1.5 years17 ± 1.4 yearsGV < 1 cm/year | (n = 18)♂174.5 ± 7.8 cm(z =-0.9 ± 1.2)♀157.7 ± 4.7 cm (z = -1.6 ± 0.8) | ♂13 cm(1.8 ± 1.0 SDs)♀9.8 cm(1.5 ± 0.8 SDs) |
| Raaijmakers 2008 | ObservationalKIGS | No | 7.5 | 10.17 | −3.24 | 34(0.17−0.77) | 0.54 | ♂ > 17 years♀ > 15 yearsGV< 2 cm/year | (n = 24)NA | 0.61 SDs |
| Noordam 2008 | Uncontrolled clinical trial | PTPN11 (22) SOS1 (1)BRAF (1) | 6.4 | 11 | −2.8 | 50 | 0.5 | 1 year after discontinuation of GHGV < 2 cm/year | (n = 29)♂171.3 cm♀151.3 cmTotal sample mean:z = -1.5 | 1.3 SDs |
| Romano 2009 | ObservationalNCGS | No | 5.6 ± 2.6 | 11.6 ± 3 | −3.5 ± 1♂-3.2♀ -3.8 | 33 ± 5 | NA | ♂: BA ≥ 16♀: BA ≥ 14(predicted in 30%) | (n = 64)♂-2♀ -2.3Total sample mean:−2.1 ± 1 | 1.4 SDs♂8.9 cm (1.2 SDs)♀ 10 cm (1.5 SDs) |
| Tamburrino 2015 | Uncontrolled clinical trial | PTPN11 (23). RAF1 (1). KRAS (1). SHOC2 (7) | 9.3 ± 4 | 6.9 ± 3.6 | −2.8 ± 0.8 | 35 | 0.53 | GV < 1 cm/year and epiphyseal closure | (n = 16)−2.2 ± 0.7 | 0.61 SDs |
| Malaquias 2019 | Retrospective longitudinal study | PTPN11 (15)Others (2) | 5.1 ± 2.0 | 11.4 ± 3.4 | −3.4 ± 0.8 | 47 | 0.4 ± 0.3 | GV < 1 cm/year for 12 months | (n = 17)♂160 cm (13)♀153.5 cm (4) (z = -2.1 ± 0.7) (17) | 1.3 ± 0.7 SDs |
| Ranke 2019 | Observational KIGS | PTPN11 (36) | 6.6 ± 2.8 | 10.4 ± 3.1 | –3.5 ± 1.0 | 36 ± 11 | ND | GV < 2 cm/year y♂: BA≥16♀: BA≥14 | (n = 140)♂ –2.1 ± 1.2 (74)♀ –2.5 ± 1.2 (66)–2.3 ± 1.2 (140) | 1.3 ± 0.7 SDs♂ 1.4 ± 0.8♀ 1.1 ± 0.7 |
BA, bone age; GH, growth hormone; GV, growth velocity; Height z, z-score for height in the reference growth charts; n, number of patients assessed at the time or near the time of reaching their final heights. NA: Not available; Δ final height: mean increase in final height calculated as the difference in z-scores in the reference growth chart and the corresponding cm value; ΔHeight z 1 y, change in z-score for height at 1 year of treatment.
Strategies aimed at decreasing activity in the RAS-MAPK pathway have attracted considerable attention, with of preclinical trials showing favourable results. Off-label use of trametinib in 2 patients with severe hypertrophic cardiomyopathy in the transplant waiting list succeeded in resolving the myocardial involvement nearly in full.39 Inhibition of the PI3/AKT/mTOR cascade with rapamycin has been proven to improve cardiac involvement in animal models of NS-ML, and the drug was effective in a critically ill patient.40
It has also been hypothesised that 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (known as statins) could be useful by inhibiting RAS farnesylation and its localization to the plasma membrane. An animal model of the use of statins in NS found increases in growth by improving chondrocyte differentiation,11 and a phase 3 clinical trial is currently underway to evaluate the efficacy of statins in improving growth in NS (ClinicalTrials.gov identifier NCT02713945).
ConclusionsNoonan syndrome is a disease with substantial heterogeneity in its clinical manifestation and underlying genetics, requiring a multidisciplinary approach to management and regular follow-up. This article presents guidelines for the follow-up of patients with NS based on the current literature. The recent authorization by the AEMPS of the use of rhGH for treatment of short stature in patients with NS adds a symptomatic treatment tool that did not use to be available in Spain.
In recent decades, advances in gene sequencing techniques have allowed the identification of the genetic cause of NS in most patients and allowed the development of many preclinical studies focused on the identification of potential therapeutic targets in these patients. Hopefully, these studies will allow the development in upcoming years of gene-targeted therapies to treat NS and other RASopathies.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Carcavilla A, Suárez-Ortega L, Rodríguez Sánchez A, et al. Síndrome de Noonan: actualización genética, clínica y de opciones terapéuticas. An Pediatr (Barc). 2020;93:61.
