



Persistent Müllerian duct syndrome (PMDS) is a disorder of sexual differentiation characterised by the persistence of müllerian derivatives (uterus and Fallopian tubes) in males with an XY karyotype and normal virilization.1
Eighty-five percent of cases of PMDS are caused by mutations in the antimüllerian hormone gene (AMH), which lead to defects in its secretion or activity, or to mutations in the gene for the type II receptor for AMH (AMHR2), which results in a clinical picture of hormonal resistance.2 It has an autosomal recessive pattern of inheritance. Measuring the levels of AMH may be useful (levels are undetectable in cases with mutations in the AMH gene or normal to high in cases with mutations in the AMHR2 gene).3,4
We present the case of a male aged 2 years and 3 months with findings compatible with Müllerian remnants during surgical correction of cryptorchidism. The patient was the product of a twin gestation that followed in vitro fertilisation and was delivered by Caesarean section at 37 weeks’ gestation with a weight of 2100g (−1.8SD) and length of 45cm (−1.85SD). The patient had bilateral cryptorchidism since birth and had developed left hydrocele by one year of life. There was no family history of interest.
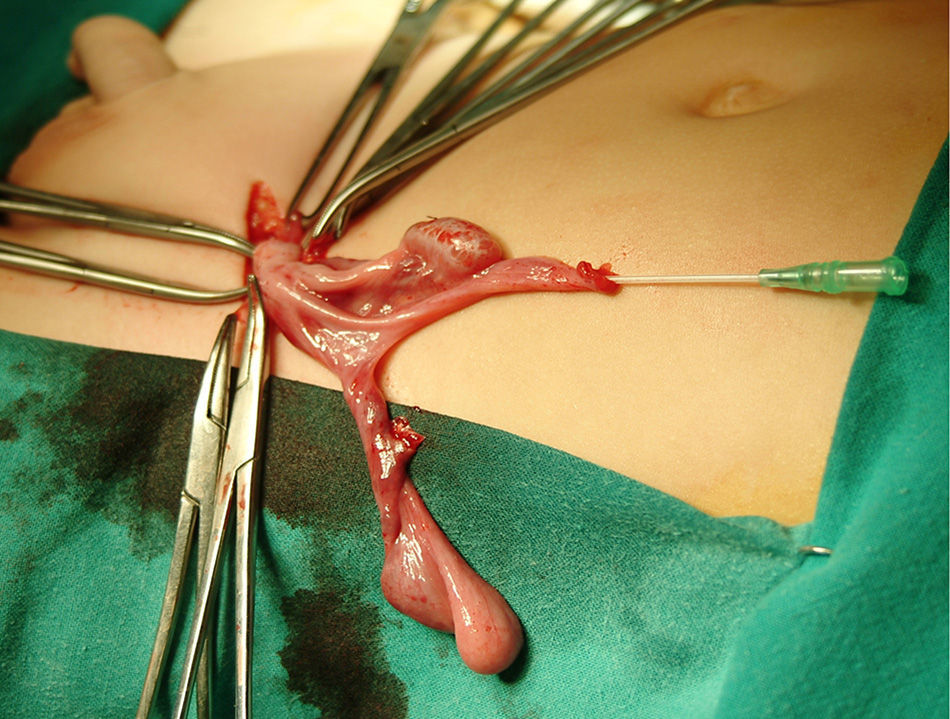
Left inguinotomy revealed a permeable duct containing gonadic tissue in proximity to a tubular structure with a permeable lumen and of salpingeal appearance. The surgical approach was expanded, revealing a contralateral gonad that was continuous with what appeared to be a hypoplastic uterus and Fallopian tubes (Fig. 1). Ureterocystoscopy allowed visualisation of a male posterior urethra with its verumontanum and seminal colliculus with normal morphology. After removal of the Müllerian remnants and biopsy of both gonads, the patient underwent left herniotomy and orchidopexy. Right orchidopexy was performed subsequently.
The pathological examination of the removed structures confirmed the presence of Müllerian duct remnants, and the two gonads were described as dysgenetic testes with a reduced mean tubular diameter and fertility index, no evidence of Leydig cells in the interstitial tissue and a normal Sertoli cell count.
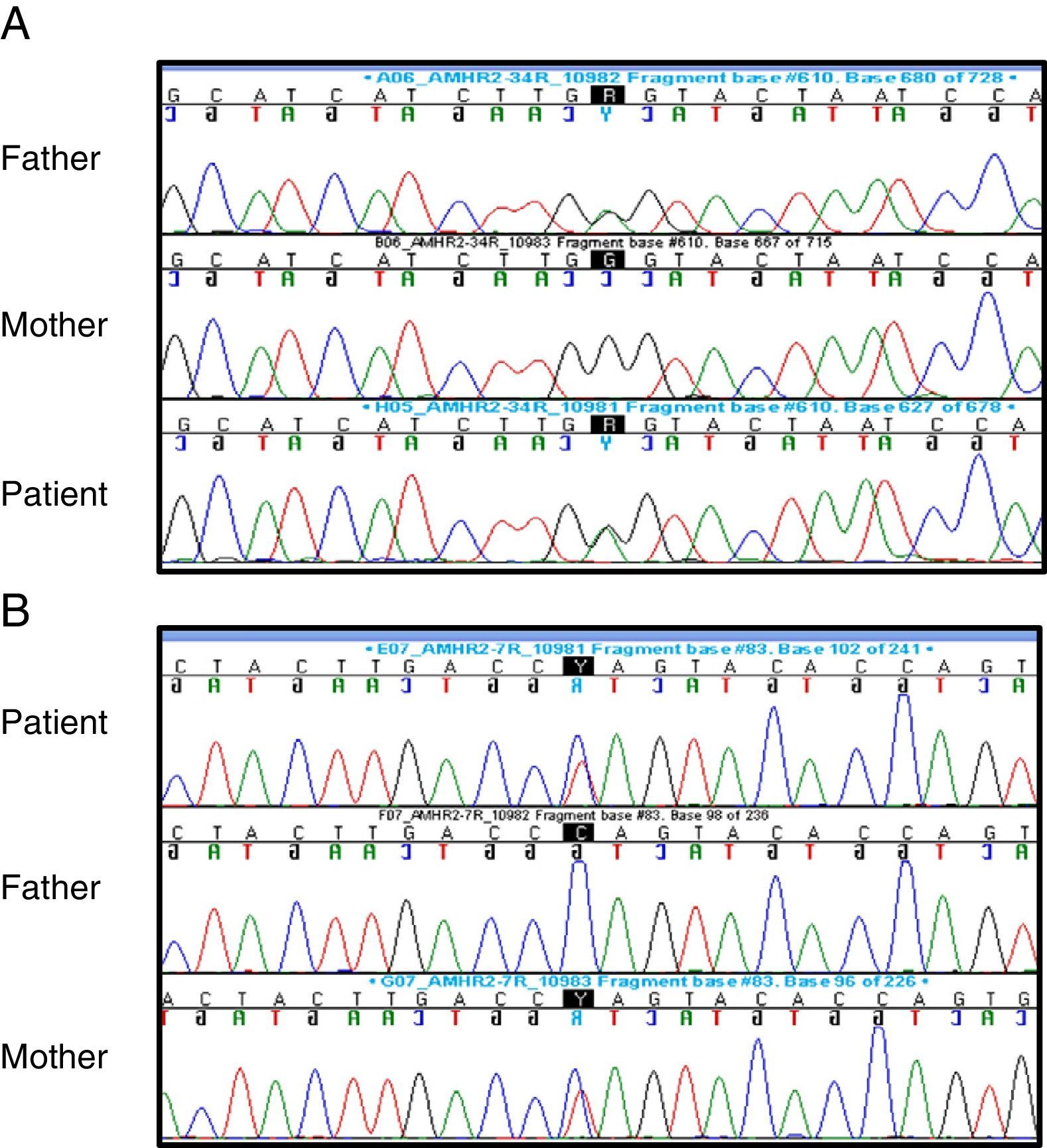
Suspicion of PMDS led to performance of the following tests: karyotype (46,XY); FSH (1.2mIU/mL; normal range, 0.2–1.4), LH (0.04ng/mL; normal range, 0.01–0.5) and testosterone (0.04ng/mL; normal range, 0.01–0.04) – baselines that were not abnormal – and AMH (816pg/L; normal range, 360–668pg/L). Sequencing of the AMHR2 gene revealed the presence of a previously described mutation in exon 4 (c.502G>A; p.Ala168Thr, rs374601719) as well as a novel mutation in exon 7 (c.877C>T; p.Gln 293*), both of which were heterozygous. The exon 4 mutation was present in the father and the exon 7 mutation in the mother, and both were heterozygous in the parents, showing that the patient was a compound heterozygote (Fig. 2).
At age 8 years and 7 months, the patient's weight and height were at the 50th percentile, his testis measured 2–3mL, his penis measured 4cm with a scrotum of normal appearance (Tanner I), and evidence of microcalcifications in the most recent checkups called for rigorous clinical and sonographic monitoring. Following performance of a LHRH test, FSH levels rose to a maximum of 5.88mIU/mL and LH levels to 3.88mIU/mL, with testosterone levels of 0.18ng/mL.
Patients with PMDS exhibit a male phenotype at birth and the diagnosis usually results from a chance finding during surgery for cryptorchidism with inguinal hernia or hydrocele.
The AMHR2 gene, located in location 12q13 of chromosome 12, comprises 8.7kb and contains 11 exons.4,5 It encodes a transmembrane membrane with an extracellular domain that binds AMH and an intracellular domain with serine/threonine kinase activity that binds and phosphorylates receptor type I, activating the transcription of target genes that mediate the action of AMH. Several mutations in this gene have been described in the literature, and the most frequent one consists of the deletion of 27 base pairs in exon 10.
The in silico analysis of the c.877C>T and c.502G>A variants using “MutationTaster” predicted that they were disease-causing. The c.877C>T mutation encodes a premature stop codon that results in a truncated protein that is missing part of the serine/threonine kinase domain. The c.502G>A substitution is located in the last base of exon 4 and seems to alter the splicing process. Furthermore, c.502G>A is a rare variant, with an allele frequency of 0.01% in the ExAC database. All of the above suggest that PMDS was due to both mutations in the AMHR2.
In PMDS, testes exhibit normal differentiation with the presence of germ cells. However, fertility may be compromised by the degeneration secondary to cryptorchidism and altered communication with the vas deferens, which in many cases are aplastic and may be disconnected from the testes due to the presence of the Müllerian derivatives.
The literature has described an increased risk of testicular tumours (15%), similar to the one described in patients with abdominal cryptorchidism5,6; this fact, along with the microcalcifications found in our patient, calls for rigorous clinical and sonographic followup.
This is an exceptional clinical case, as few cases of patients with this disease have been described in the international literature.
Please cite this article as: Orós-Millán ME, Muñoz-Calvo MT, Nishi MY, Mendonca BB, Argente J. Síndrome de persistencia del conducto de Müller debido a mutación en el gen del receptor de la hormona antimülleriana (AMHR2). An Pediatr (Barc). 2017;86:94–95.
