










Although primary immune thrombocytopenia (ITP) is rare in childhood, it is the most frequent cause of thrombocytopenia. There have been attempts to establish risk factors to predict the progression of the disease in order to optimise its management, which has changed in recent years due to, among other reasons, specialised care.
Material and methodsA retrospective, observational and analytical study was conducted on patients diagnosed with ITP over a 3-year period in a Paediatric Haematology specialist clinic.
ResultsFrom the epidemiological, clinical and analytical point of view, the characteristics of this group are similar to others. Most of the patients (23/31, 74.2 %) had ITP for less than 12 months, with there being no serious complications related to the disease or the treatment received. It was established that risk factors were related to being slowly evolving (lower event-free survival (EFS)) with no statistical significance, female gender, age over 10 years, leukopenia absence of initial severe thrombocytopenia, and non-specialised care. The absence of a history of infection was significantly related to a lower EFS.
ConclusionsThe epidemiological and analytical risk factors for a slowly evolving ITP are the same that described in the literature. Patients treated before the beginning of specialised care also had a lower EFS. These data seem to support the current recommendation that rare diseases should be managed in specialised units.
La trombopenia inmune primaria (PTI) es poco frecuente en la infancia, pero es la causa más habitual de trombopenia. Se han intentado establecer factores de riesgo para predecir su evolución, con el objetivo de poder optimizar su manejo, que se ha modificado en los últimos años, debido entre otros factores, a una atención más especializada.
Material y métodosEstudio retrospectivo, observacional y analítico de los pacientes con PTI, en un periodo de 3 años, en una consulta especializada en Hematología Pediátrica.
ResultadosDesde el punto de vista epidemiológico, clínico y analítico, las características de esta serie son similares a las de otros grupos. La mayoría de los pacientes (23/31; 74.2 %) presentaron una PTI de duración menor de 12 meses, sin complicaciones graves relacionadas con la enfermedad ni con el tratamiento. Se establecieron como factores de riesgo relacionados con una evolución tórpida (supervivencia libre de eventos (SLE) menor), sin alcanzar la significación estadística, el sexo femenino, la edad mayor de 10 años, la leucopenia, la ausencia de trombopenia grave inicial y la atención no especializada. La ausencia de antecedente de infección se relacionó significativamente con una SLE menor.
ConclusionesLos factores de riesgo de evolución tórpida de PTI epidemiológicos y analíticos de este estudio coinciden con los descritos en la literatura. Presentaron una SLE menor los pacientes tratados antes del inicio de la atención especializada. Estos datos parecen apoyar la recomendación actual de que las enfermedades poco frecuentes, como ésta, se controlen en unidades especializadas.
Primary immune thrombocytopenia (PIT) is a disease characterised by an isolated decrease in the platelet count to under 100 000/mm3 without an identified trigger. At present, the qualifier “chronic” is applied starting from 12 months post diagnosis.1,2
It is the most common form of acute thrombocytopenia in healthy children, but it is not a frequent disease. The incidence in children aged less than 15 years is estimated at about 5 cases per 100 000 inhabitants per year.3
It is mainly characterised by haemorrhagic manifestations at 3 levels: the skin, mucosae and internal organs. Most patients are asymptomatic or have cutaneous/mucosal manifestations. Different clinical scales have been developed to objectively assess the intensity of bleeding, among which one of the most widely used is the one proposed by the World Health Organization (WHO): grade 0: no evidence of bleeding; grade 1: minor bleeding (petechiae, ecchymoses, mucocutaneous haemorrhage, retinal bleeding without visual impairment); grade 2: gross bleeding (melanotic stool, haematemesis, haematuria, haemoptysis); grade 3: any bleeding requiring red blood cell (RBC) transfusion; grade 4: retinal bleeding with visual impairment, central nervous system bleeding. Only 3 % of paediatric patients develop clinically significant bleeding, and the most frequent manifestations in this subset is epistaxis and gastrointestinal bleeding.3 The intensity of bleeding appears to be associated with the severity of thrombocytopenia, although there is no exact or direct correlation between these 2 variables. The most severe complication is intracranial haemorrhage (ICH), with a frequency that ranges from 0.1 % to 0.6 %. To date, no clear predictor of ICH has been identified in these patients.3–6
Primary immune thrombocytopenia is a diagnosis of exclusion. The key elements for diagnosis of PIT are an unremarkable personal and family history and a normal physical examination (with the exception of bleeding manifestations) associated with the presence of isolated thrombocytopenia. Most patients have a history of viral infection (60 % of upper respiratory tract infection and exanthema), vaccination (most frequently the measles-mumps-rubella [MMR] vaccine) or bacterial infection in the weeks preceding onset.3,4 Any abnormalities in the blood count other than isolated thrombocytopenia and/or abnormalities in the physical examination (dysmorphic features, visceromegaly etc) or in the clinical history (recurrent infection, fever, medication, etc) should at least prompt reconsideration of the PIT diagnosis.7,8
Primary immune thrombocytopenia is a benign and self-limiting disease. Different registers and published studies show remission in 60 % of cases within 6 months of diagnosis, independently of the treatment given.3,9 The identification of predictors of remission has proven challenging. There is evidence of an association between a favourable outcome and the following variables: male sex, age less than 10 years, abrupt onset, history of infection preceding onset and a very low platelet count (<5000/mm3).10–13 The presence of leukopenia also seems to be associated with an increased risk of persistent or chronic PIT.14
The drugs used most frequently in the management of PIT have been corticosteroids and intravenous immunoglobulin (IVIG).15–18 Historically, treatment was prescribed with the aim of increasing the platelet counts. In light of the rarity of significant bleeding, the lack of evidence demonstrating that pharmacotherapy prevents severe bleeding and the economic cost and known toxicity of the different drugs available, the approach to the management of PIT is more conservative, and observation is currently recommended for asymptomatic or mildly symptomatic children. When pharmacotherapy is used, since there is no evidence of the superiority of any of the available drugs, it is recommended that the advantages and disadvantages of each are considered and discussed with the patients and their families to establish an individualised treatment plan.3,9,19 This shift in the approach to the management of PIT has been reflected in the different protocols and guidelines published in recent years, including the protocol of the Sociedad Española de Hematología y Oncología Pediátricas (Spanish Society of Paediatric Haematology and Oncology, SEHOP).19–21
In 2013, a specialised clinic in paediatric haematology opened its doors in the Hospital Infantil Miguel Servet of Zaragoza, Spain, to carry out the diagnosis, treatment and follow-up of patients with diagnosis of noncancerous blood disorders, including PIT. Until then, cases of PIT in this children’s hospital were managed by various providers that were not specialists in paediatric haematology. Once the clinic opened, it routinely applied the protocols endorsed by paediatric haematology societies.20
The aim of our study was to establish the clinical and laboratory characteristics and the outcomes of patients with PIT evaluated in this specialty clinic and to assess the risk factors that may be associated with a protracted course.
Sample and methodsWe conducted a retrospective, observational and analytical study by reviewing the health records of patients with a diagnosis of PIT managed between January 2013 and October 2016 in the paediatric haematology clinic of the Hospital Infantil Miguel Servet in Zaragoza.
We included patients aged less than 15 years with a diagnosis of PIT managed in the clinic during the period under study regardless of the date of the initial diagnosis. We excluded patients with insufficient follow-up data.
We applied the diagnostic criteria, definitions for assessment of response to treatment and the treatments specified in the current protocol of the SEHOP for the management of PIT.20
We collected data on demographic and epidemiologic variables, laboratory tests, treatment, response to treatment, follow-up and outcomes. The data collection process adhered to the current legislation on the protection of personal data.
We compared variables by means of the χ² and Student t tests. To assess the association between variables, we performed binary logistic regression analyses where the dependent variable was the type of PIT based on the duration of illness (less than or greater than 12 months). We also performed a survival analysis, defining event-free survival (EFS) as the time elapsed (in months) from diagnosis to whichever of the following occurred first: low platelet count of less than 30 000/mm3, death, discharge or end of the study (October 31, 2016). We plotted survival curves using the Kaplan-Meier method. We compared the difference in EFS based on different clinical, diagnostic and treatment factors (age, sex, presence of mucosal bleeding, previous infection, platelet and white blood cell counts, management in the specialty clinic or prior to the opening of the speciality clinic) with the Kaplan-Meier method and the long-rank test. We defined statistical significance as a p-value or B value of less than 0.05. The statistical analysis was performed with the software IBM SPSS version 24.
ResultsBetween January 2013 and October 2016, 509 patients were assessed in the paediatric haematology clinic for some form of noncancerous blood disorders. Of this total, 44 (8.6 %) had a diagnosis of thrombocytopenia, and 31 (70 % of patients with thrombocytopenia) a diagnosis of PIT. Based on this finding and the census data of the Instituto Aragonés de Estadística (Institute of Statistics of Aragon), we calculated an incidence in children aged less than 15 years of 2.6 cases per 100 000 inhabitants per year.
The distribution of patients by sex was 18 male and 13 female (1.3:1 ratio). The mean age at diagnosis was 4.5 years, with a median of 4 years and a range of 0–13 years. Most patients (27/31) were aged less than 10 years at the time of diagnosis.
When it came to the clinical manifestations, 2 patients (6.5 %) did not have any overt bleeding, and the diagnosis was made when blood tests were done for other reasons. In the remaining 29 patients there were bleeding manifestations: cutaneous in all, as well as mucosal in 12 (38.7 %), and epistaxis was the most frequent form of bleeding (10/12; 83 %), followed by gastrointestinal bleeding. We did not find a recent history of vaccination in any of the patients, but found a history of recent infection in 13 (41.9 %).
The mean platelet count at diagnosis was 11 000/mm3, and the median was 9000/mm3. In addition, 83.8 % of the patients had platelet counts of less than 20 000/mm3 at diagnosis and 13 % had very severe thrombocytopenia (< 5000 platelets/mm3). The platelet count at admission did not exceed 50 000/mm3 in any case. The initial haemoglobin concentration was abnormal in 2 patients, who underwent bone marrow analysis, confirming the peripheral mechanism of the disease. Six patients (19.4 %) had white blood cell (WBC) counts of less than 6000 cells/mm3 at diagnosis, and most of them (66 %) progressed to chronic PIT (Table 1).
Of the 31 patients, 3 (9.7 %) did not receive any pharmacotherapy during the follow-up, and the thrombopenia resolved within 6 months in all. Thirteen (46.8 %) received 1 treatment, most frequently prednisone (8 patients), followed by IVIG (5 patients). Twelve patients required 2 treatments, either due to poor clinical response and/or poor improvement of laboratory parameters, and treatments other than prednisone or IVIG were used in 3 patients (dexamethasone, rituximab, thrombopoietin receptor agonists, splenectomy). One patient had haemodynamically significant epistaxis requiring transfusion. None of the patients died or had life-threatening haemorrhage in the period under study. Using the bleeding scale of the WHO, most patients (28/31; 90.3 %) had grade 1–2 bleeding. There were 2 cases of grade 0, 1 case of grade 3 and no cases of grade 4 bleeding.
Twenty-nine patients (93.5 %) reached platelet counts greater than 100 000/mm3 after a mean of 150 days and a median of 65 days. Eleven (35.4 %) experienced a decrease of platelets to less than 30 000/mm3 during the follow-up, with a mean time elapsed from diagnosis of 87 days and a median of 30 days. All patients that experienced a decrease in the platelet count received at least 1 treatment at some point: 3 patients prednisone alone, 4 prednisone and IVIG and 4 at least 3 immunomodulating agents.
In most of the sample, PIT resolved within 12 months of diagnosis (23/31; 74.2 %), while 8 patients (25.8 %) eventually met the criteria for chronic PIT. In the group that developed chronic PIT, there was a predominance of female patients, the mean age was greater compared to the total sample, the mean platelet count at diagnosis was higher, the mean WBC count at diagnosis was lower, and there was no history of recent infection. Compared to patients with acute PIT or recurrent PIT of less than 12 months’ duration, we found that development of chronic PIT was significantly associated with greater age at diagnosis, the presence of leukopenia and the absence of previous infection. We did not find a statistically significant association with the platelet count, the presence of mucosal bleeding or the treatment received (Tables 2 and 3).
Clinical, laboratory and treatment characteristics of patients by duration of disease.
| All patients (n = 31) | ≤ 12 months (n = 23) | > 12 months (n = 8) | |
|---|---|---|---|
| Sex, n (%) | |||
| Male | 18 (58.1) | 15 (65.2) | 3 (37.5) |
| Female | 13 (41.9) | 8 (34.8) | 5 (62.5) |
| Age in years | |||
| Mean (range) | 4.5 (0–13) | 3.7 (0–13) | 7 (4–11) |
| <10 years, n (%) | 27 (87.1) | 22 (95.7) | 5 (62.5) |
| ≥10 years, n (%) | 4 (12.9) | 1 (4.3) | 3 (32.5) |
| Cutaneous bleeding, n (%) | |||
| No | 2 (6.5) | 2 (8.7) | 0 |
| Yes | 29 (93.5) | 21 (91.3) | 8 (100) |
| Mucosal bleeding, n (%) | |||
| No | 19 (61.3) | 14 (60.9) | 5 (62.5) |
| Yes | 12 (38.7) | 9 (39.1) | 3 (37.5) |
| Previous infection, n (%) | |||
| No | 18 (58.1) | 10 (43.5) | 8 (100) |
| Yes | 13 (41.9) | 13 (56.5) | 0 |
| Platelet count in sample | |||
| Mean/mm3 | 11 079 | 9889 | 14 500 |
| Median/mm3 | 9000 | 9000 | 9000 |
| Range/mm3 | 457–47 000 | 457–27 000 | 5000–47 000 |
| Platelet count, n (%) | |||
| <10 000/mm3 | 16 (51.6) | 12 (52.2) | 4 (50) |
| ≥10 000/mm3 | 15 (48.4) | 11 (47.8) | 4 (50) |
| Platelet count, n (%) | |||
| <5000/mm3 | 4 (12.9) | 4 (17.4) | 0 |
| ≥5000/mm3 | 27 (87.1) | 19 (89.6) | 8 (100) |
| WBC count in sample | |||
| Mean | 9891 | 10 560 | 7323 |
| Median | 10 100 | 10 700 | 7045 |
| Range | 4900–15 700 | 4900–15 700 | 4900–11 150 |
| WBC count, n (%) | |||
| <6000/mm3 | 6 (19.4) | 2 (8.7) | 4 (50) |
| ≥6000/mm3 | 25 (80.6) | 21 (91.3) | 4 (50) |
| Treatment, n (%) | |||
| No | 3 (9.7) | 3 (13) | 0 |
| Yes | 28 (90.3) | 20 (87) | 8 (100) |
| Type of treatment, n (%) | |||
| IVIG alone | 5 (16.1) | 5 (21.7) | 0 |
| Prednisone alone | 8 (25.8) | 5 (21.7) | 3 (37.5) |
| IVIG + prednisone | 12 (38.7) | 10 (43.5) | 2 (25) |
| IVIG + prednisone + other | 3 (9.7) | 0 | 3 (37.5) |
| Splenectomy | 3 (9.7) | 1 (4.3) | 2 (25) |
IVIG, intravenous immunoglobulin; WBC, white blood cell.
Variables with a statistically significant association with chronic primary immune thrombocytopenia (duration > 12 months).
| ≤12 months (n = 23) | >12 months (n = 8) | P | |
|---|---|---|---|
| Age in years | |||
| Mean | 3.70 | 7 | .01 |
| <10 years | 22 | 5 | .01 |
| ≥10 years | 1 | 3 | |
| Total WBC count | |||
| Mean | 10 560 | 7323 | .01 |
| < 6000/mm3 | 2 | 4 | .01 |
| ≥ 6000/mm3 | 21 | 4 | |
| Previous infection | |||
| No | 10 | 8 | .005 |
| Yes | 13 | 0 | |
WBC, white blood cell.
As for the type of care received by patients with PIT, we found that in 13 cases the diagnosis was made before the haematology clinic opened, while the remaining 18 received the diagnosis when the clinic was already active.
The binary logistic regression analysis in which the type of PIT (acute/persistent or chronic) was the dependent variable did not find significant associations with any of the independent variables: sex, age, WBC and platelet counts at diagnosis, presence of mucosal bleeding or treatment received.
We compared the EFS based on different epidemiological, clinical and treatment factors and found lower values (although the differences were not statistically significant) in female patients (P = .37), patients aged more than 10 years (P = .056), without evidence of mucosal bleeding (P = .95), with platelet counts greater than 5000/mm3 (P = .16), with a low WBC count of less than 6000/mm3 (P = .06), and treated before the specialised haematology clinic opened (P = .06). The EFS was significantly lower (P = .001) in patients without a previous history of infection (Table 4 and Figs. 1–4).
Event-free survival based on demographic, clinical and laboratory variables and management period.
| Variable | Patients (n) | Events (n) | EFS (%) | P |
|---|---|---|---|---|
| Sex | 30 | 11 | ||
| Male | 17 | 5 | 70. 6 | .37 |
| Female | 13 | 6 | 56.8 | |
| Age | 30 | 11 | ||
| <10 years | 26 | 8 | 69.2 | .056 |
| ≥10 years | 3 | 3 | 25 | |
| Mucosal bleeding | 30 | 11 | ||
| No | 18 | 7 | 61.1 | .95 |
| Yes | 12 | 4 | 66.7 | |
| Previous infection | 30 | 11 | ||
| No | 17 | 11 | 35 | <.05 |
| Yes | 13 | 0 | 100 | |
| Platelet count | 30 | 11 | ||
| < 5000/mm3 | 4 | 0 | 100 | .16 |
| ≥ 5000/mm3 | 26 | 11 | 57.7 | |
| WBC count | 30 | 11 | ||
| <6000/mm3 | 6 | 4 | 33.3 | .06 |
| ≥6000/mm3 | 24 | 7 | 70.8 | |
| Management period | 30 | 11 | ||
| Before opening of specialty clinic | 20 | 10 | 50 | .06 |
| Specialty clinic | 10 | 1 | 90 | |
EFS, event-free survival; WBC, white blood cell.
The p-values correspond to the log-rank test.
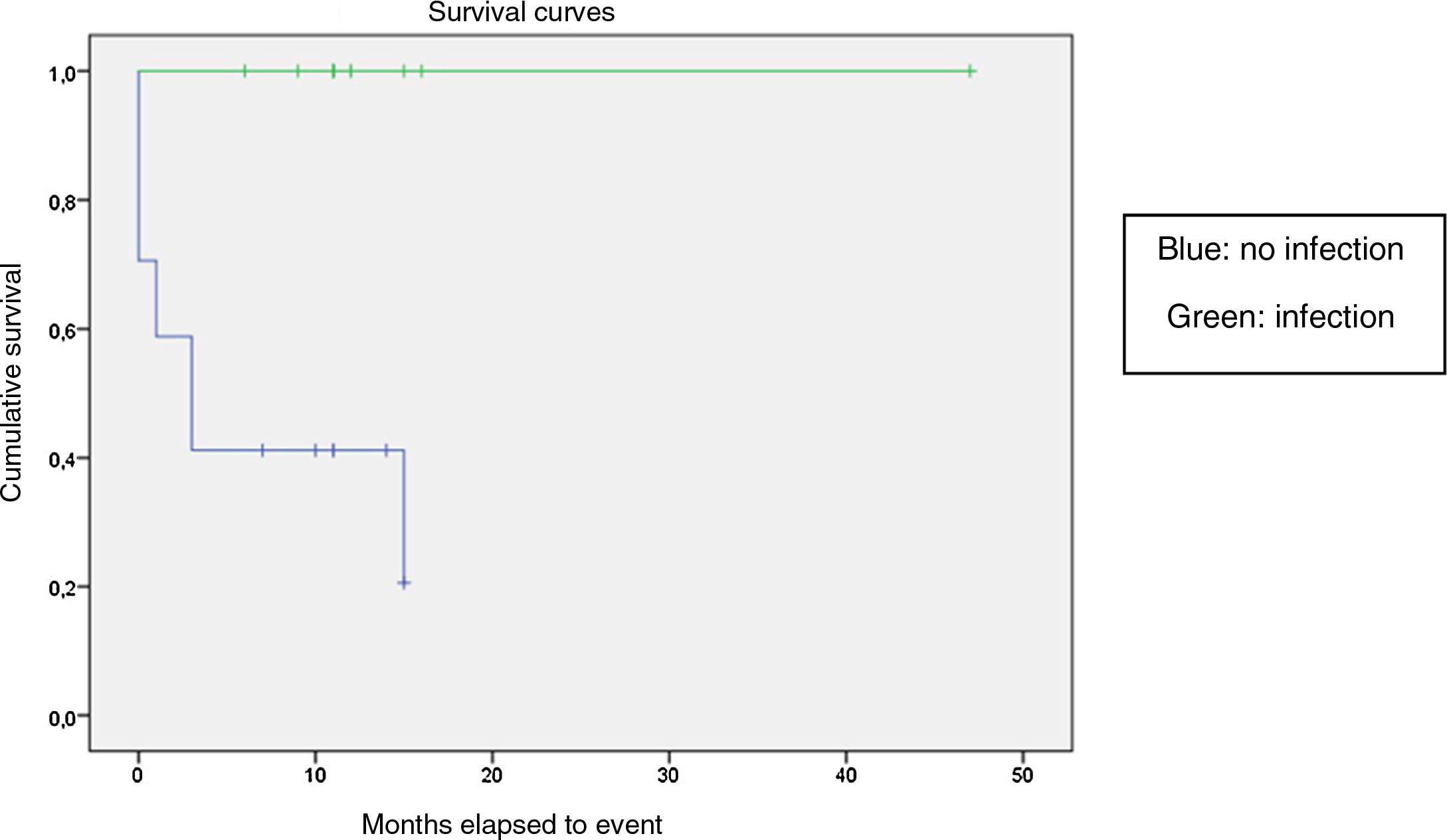
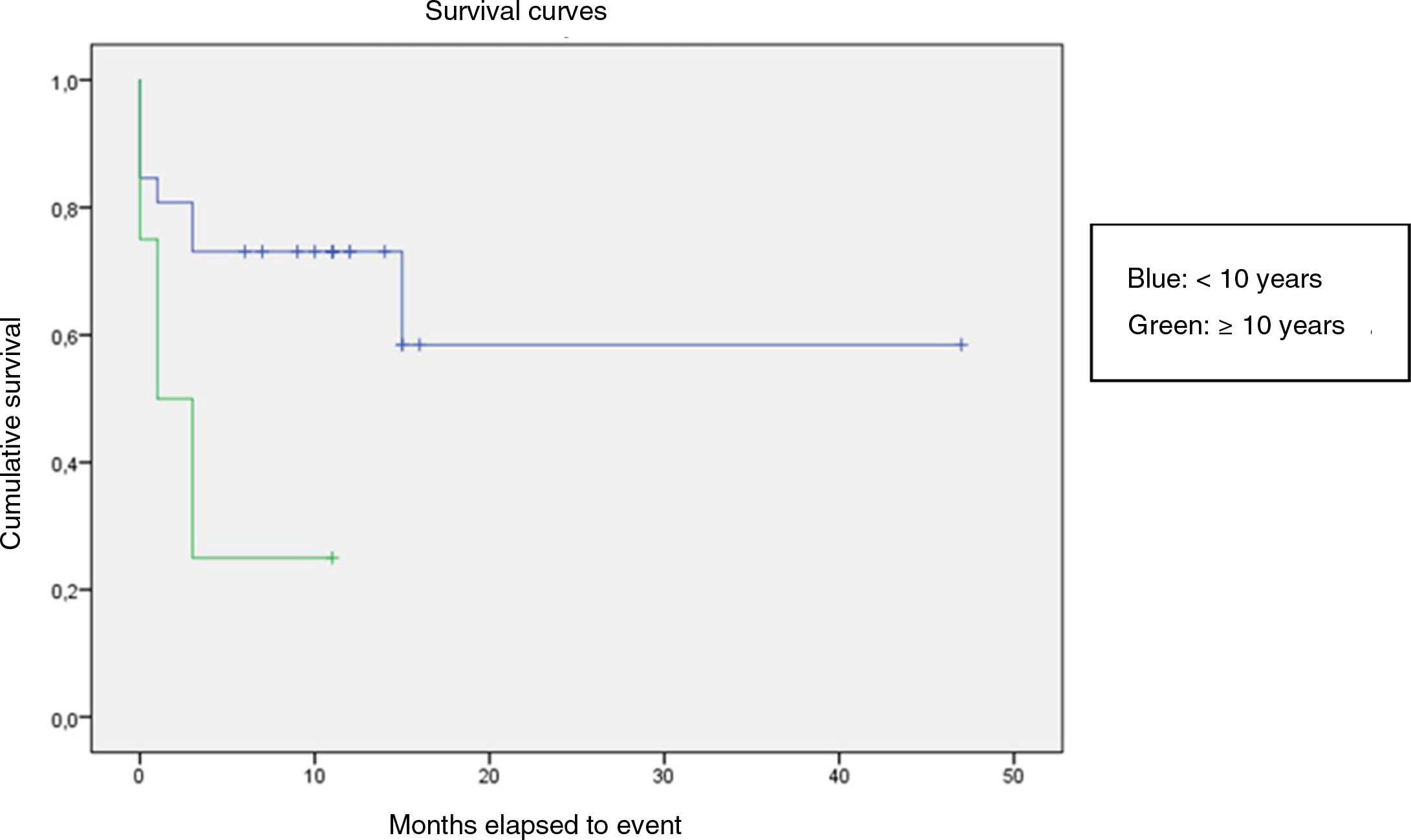
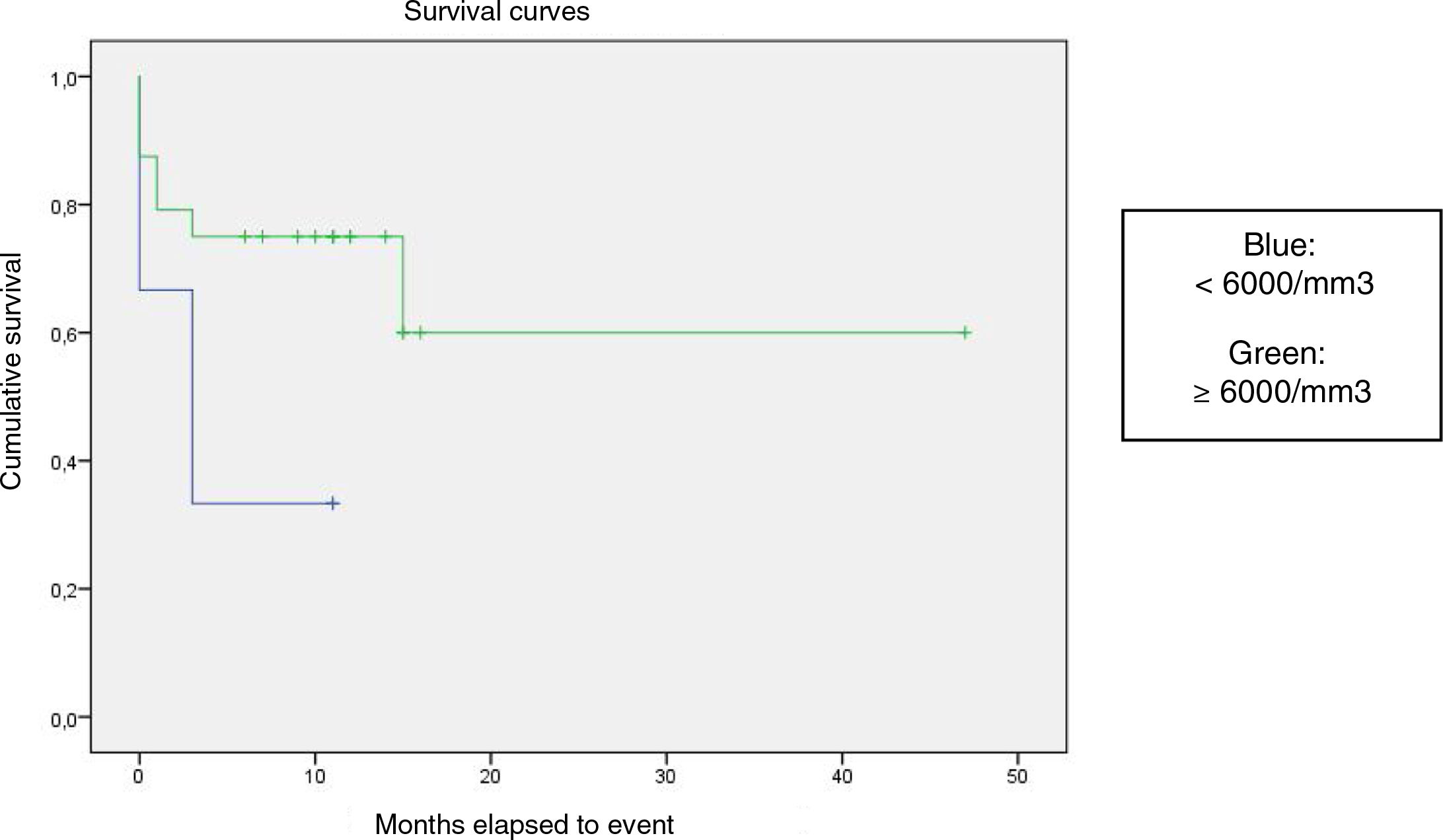
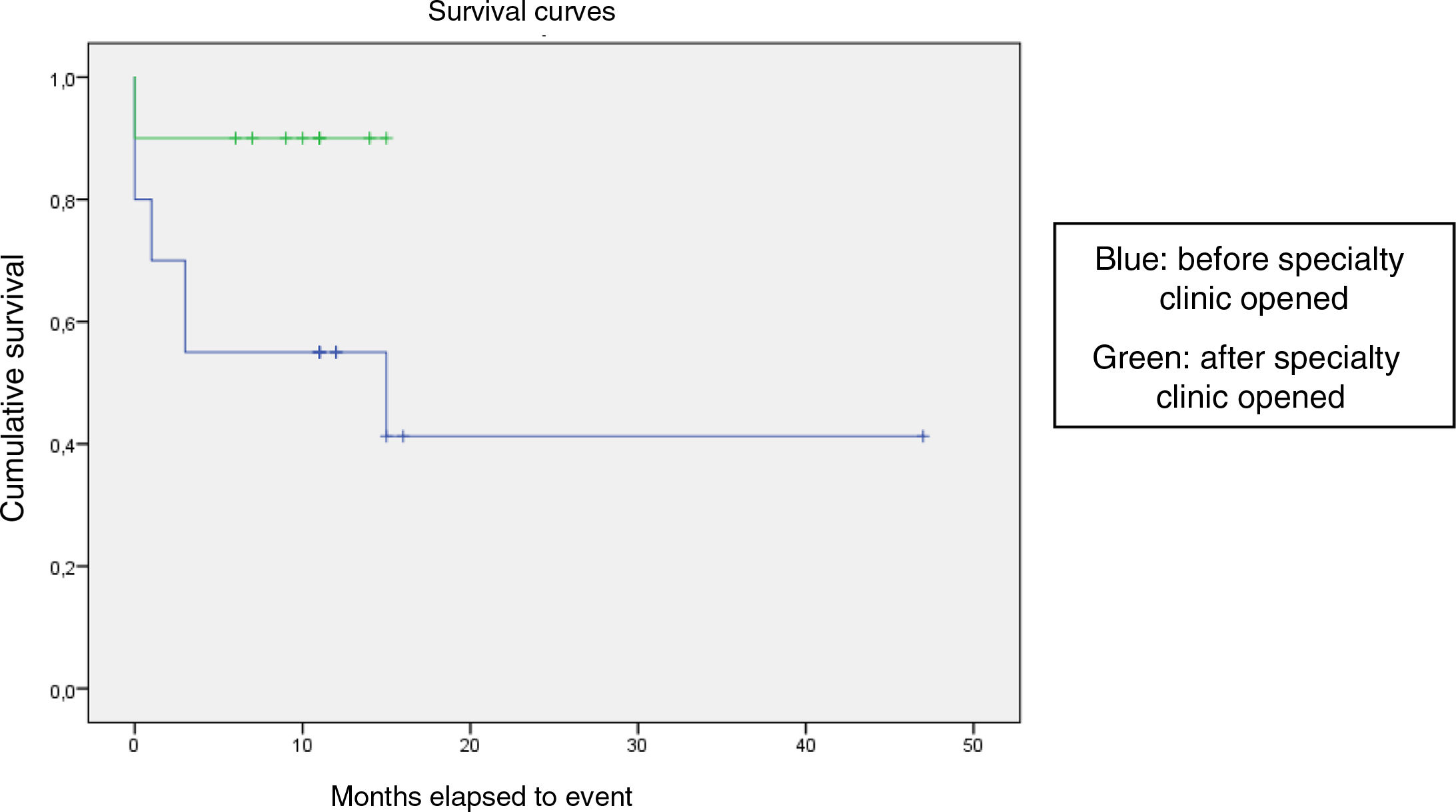
Kaplan-Meier curve of the event-free survival (EFS) based on the management period (P = .06).
PARCHEOS: cambiar las comas en los decimales a puntos en el eje vertical.
Cumulative survival
Months elapsed to event
Survival curves
Blue: before specialty clinic opened
Green: after specialty clinic opened
Of the 31 patients under study, 19 (61.3 %) were discharged from the clinic after at least 1 year of follow-up past the resolution of thrombocytopenia. Twelve patients continued in follow-up by the paediatric haematology clinic due to either chronic disease or because a year of follow-up had not been completed at the end of the study period.
DiscussionThe epidemiological data obtained in our study were consistent with the previous literature and the data collected in various registers as pertains the age and sex distribution of cases of PIT, with the highest proportion corresponding to male patients aged 2–4 years.3,5 The estimated incidence rate based on our findings of 2.6 cases per 100 000 paediatric patients per year was lower than the 5 cases per 100 000 per year reported in most previous studies.3,5 This lower estimate may be attributed to several factors: first, that not all cases of PIT in Aragon were managed in the hospital where we conducted the study, and also that in our case series, most patients had severe thrombocytopenia (mean platelet count of 11 000/mm3, maximum count of 47 000/mm3), and that the study did not include patients with mild thrombocytopenia that may have met the diagnostic criteria for PIT. Had we included these patients with milder manifestations, the incidence in our study would probably have been closer to the one reported by other authors.3,5
The frequency of clinically significant bleeding manifestations (3 %) was consistent with the literature, and the frequency of severe bleeding has even lower than described in previous studies.3,6,9 There were no cases of life-threatening bleeding during the follow-up. Two children developed anaemia in association with the bleeding episodes, of who only 1 required transfusion of blood products. These findings support the notion that PIT is a benign disease in children that is rarely associated with significant morbidity or mortality.
Primary immune thrombocytopenia is frequently a self-limiting disease lasting a few weeks or months, but in approximately 25%–30% of cases it becomes chronic.3,13,14 In our study, most samples achieved normal platelet counts in less than 6 months, and more than two thirds of the sample had a disease duration of less than 12 months.
We did not find differences based on the treatment received. The most salient finding was that the 3 patients that did not receive any treatment recovered spontaneously in less than 6 months. The interpretation of these findings is limited by the small sample size, but they seem to corroborate that the outcome of PIT (resolution versus chronic disease) does not depend on the treatment given.3,10
The identification of factors that may influence the outcome of PIT has become a key objective in the research devoted to this disease,9–14 as clinicians facing a newly diagnosed case cannot determine whether the child has a disease that is self-limiting or that will become a chronic ailment, a difference that has a significant impact on the management and the environment of the patient.
In the sample under study, patients with chronic PIT were more frequently female, had a higher mean age and presented with nonsevere thrombocytopenia and leukopenia and did not report recent infection in the history taking at the time of diagnosis. We found a statistically significant association of chronic PIT with older age, the presence of leukopenia and the absence of recent infection.
The regression analysis did not yield statistically significant results, probably due to the small sample size, but the survival analysis confirmed the absence of a history of recent infection as a statistically significant risk factor for recurrence. A possible explanation is that in case there is a clear infectious trigger of the autoimmune process, the thrombocytopenia would exhibit the expected, classic course, self-limiting and benign. The absence of such an infectious trigger could suggest that the pathophysiological process underlying the immune dysregulation is multifactorial and more complex, with a greater likelihood of development of chronic disease. Other factors that were associated with a lower EFS, with results that were not statistically significant but neared the significance threshold, were older age, the presence of leukopenia and management in a setting other than the specialised haematology clinic.
ConclusionsThe epidemiological and outcome findings in our sample were consistent with previous data: most patients were male, of preschool age, had a history of recent infection and most commonly had bleeding manifestations at the skin level, severe thrombocytopenia at diagnosis and a favourable outcome with resolution of PIT in less than 12 months.
Patients who are female, school-aged or older, without evidence of related previous infection, without signs of mucosal bleeding or with nonsevere thrombocytopenia or leukopenia at diagnosis seem to be at higher risk of protracted and recurrent disease with a tendency toward chronic PIT. Given the low prevalence of this disease, performance of a prospective multicentre study with a larger sample would be useful to corroborate these findings with greater accuracy and statistical significance.
In our sample, patient outcomes improved once the specialised paediatric haematology clinic was established. Based on our findings, albeit taking into consideration the limitations of our study, it seems reasonable to recommend that relatively infrequent diseases, such as PIT, be managed in specialised units with the aim of minimising heterogeneity in clinical management and offer the most appropriate treatment available based on up-to-date and high-quality scientific evidence.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Rodríguez-Vigil Iturrate C, et al. Trombocitopenia inmune primaria: experiencia de una consulta especializada en Hematología. An Pediatr (Barc). 2020;93:16–23.
