






Pulmonary hypoplasia is the most frequent congenital anomaly associated with perinatal mortality.
Material and methodsA retrospective and descriptive review was conducted on cases of patients diagnosed with pulmonary hypoplasia between 1995 and 2014 in a tertiary university hospital. An analysis was made of the prenatal imaging, clinical manifestations, post-natal diagnostic tests, treatment and management, long-term follow up, and survival data.
ResultsA total of 60 cases were identified, all of them with prenatal imaging. Sixteen patients required foetal surgery. Congenital diaphragmatic hernia was the most frequent diagnosis. Main clinical presentation was respiratory distress with severe hypoxaemia and high requirements of mechanical ventilation. Mortality rate was 47% within first 60 days of life, and 75% for the first day of life. Pneumonia and recurrent bronchitis episodes were observed during follow-up. They had a lung function obstructive pattern, and their quality of life and exercise tolerance was good.
ConclusionsHigh neonatal mortality and significant long-term morbidity associated with pulmonary hypoplasia requires an early diagnosis and a specialised multidisciplinary team management.
La hipoplasia pulmonar es la anomalía congénita más frecuentemente asociada a mortalidad perinatal.
Material y métodosSe ha realizado un estudio descriptivo retrospectivo de los casos de hipoplasia pulmonar diagnosticados entre 1995 y 2014 en un hospital universitario de tercer nivel, analizando estudios prenatales, manifestaciones clínicas, pruebas diagnósticas, tratamiento, datos de seguimiento a largo plazo y supervivencia.
ResultadosSe identificaron 60 casos, todos con estudio prenatal. Dieciséis recibieron intervención quirúrgica intraútero. La hernia diafragmática congénita fue la entidad más frecuentemente asociada. La manifestación clínica más habitual fue distrés respiratorio neonatal, hipoxemia grave y necesidad de soporte con ventilación mecánica. Se halló un 47% de mortalidad antes de los 60 días de vida y del 75% en las primeras 24 h de vida. Durante el seguimiento de los supervivientes se detectaron episodios de neumonías y bronquitis recidivantes, función pulmonar con patrón obstructivo y aceptable calidad de vida y tolerancia al ejercicio.
ConclusionesLa elevada mortalidad neonatal y la importante morbilidad a largo plazo de la hipoplasia pulmonar requieren de un diagnóstico temprano y la intervención de un equipo multidisciplinar especializado.
Pulmonary hypoplasia (PH) is a congenital anomaly characterised by impaired growth and development of the lung parenchyma, airways and vessels.1–5 Its incidence in the general population is of nine to eleven cases per 10,000 live births,2,3 although this must be an underestimate since infants with lesser degrees of PH survive the neonatal period.2 The prevalence of PH reported in perinatal autopsy case series ranges between 7.8% and 26%,1,3 and it is the anomaly most frequently associated with perinatal mortality.1–6
Pulmonary hypoplasia most frequently develops secondary to abnormalities in the thoracic cavity, foetal breathing movements, foetal lung liquid at positive pressure and volume of amniotic fluid.1–7Table 1 summarises and enumerates the underlying conditions most commonly associated to PH.4,8,9 The clinical manifestations depend on the degree of lung involvement, ranging from severe bilateral forms to milder unilateral or lobar forms.1,10 During the neonatal period, it can manifest with respiratory insufficiency, pulmonary hypertension or pulmonary haemorrhage. It characteristically presents with respiratory insufficiency of sudden onset that requires mechanical ventilation with high ventilatory pressures in the absence of atelectasis or obstruction.2,11
Classification of the underlying conditions that may cause secondary pulmonary hypoplasia.
| Abnormalities of the thoracic cavity | Abnormalities in foetal lung fluid flow | Abnormalities in foetal breathing movements |
|---|---|---|
| Space-occupying lesions in the chest Congenital diaphragmatic hernia Malformation of the airways (such as cystic adenomatoid malformation) Pleural effusion Exomphalos Gastroschisis | Hypoplastic left or right heart Pulmonary stenosis Oligohydramnios Renal agenesis Cystic dysplasia Urinary outflow obstruction Prolonged rupture of membranes Hydrops fetalis | Neuromuscular disorders Neurological problems Pena-Shokeir syndrome |
| Chest wall malformations | ||
| Kyphoscoliosis | ||
| Skeletal dysplasias |
The diagnosis can be suspected prenatally based on measurements of lung volume by means of three-dimensional (3D) ultrasound and magnetic resonance imaging (MRI).12,13 The diagnostic tests used after birth are thoracic computed tomography (CT) and lung scintigraphy, and the diagnosis is confirmed by pathologic examination.1–4
Different surgical approaches are used prenatally to attenuate the severity of PH in the foetus, such as drainage in cases of hydrothorax or foetoscopic tracheal occlusion (FETO) in severe cases of congenital diaphragmatic hernia.14,15 After birth, the management of PH consists of supportive measures to guarantee adequate oxygenation during lung growth and development.
The established approach to the postnatal management of congenital diaphragmatic hernia is surgical repair followed by the use of invasive ventilatory support techniques with permissive hypercapnia combined with aggressive treatment for pulmonary hypertension.10,15–18
Recent studies have been pursuing the optimisation of the diagnostic methods, treatments and postnatal care used in patients with PH.19 The aim of our study was to describe the aetiology, diagnostic methods and clinical outcomes of cases of PH diagnosed in a tertiary university hospital.
Materials and methodsWe conducted a descriptive retrospective study of patients with PH diagnosed between 1995 and 2014 at the Hospital Universitario Vall d’Hebron de Barcelona that received care in the departments of obstetrics, neonatology, paediatric surgery and paediatric pulmonology.
We analysed the following variables: gestational age of the foetus at diagnosis, prenatal ultrasound abnormalities, prenatal surgery, gestational age at birth, age of onset of respiratory symptoms, clinical manifestations, aetiological diagnosis, diagnostic tests used, pathological findings, laterality of lung involvement, duration of required mechanical ventilation and oxygen supplementation, clinical respiratory outcomes (presence or absence of recurrent episodes of wheezing or infection), pulmonary function outcomes, quality of life and survival.
We studied the following prenatal surgical interventions: insertion of pleural-amniotic shunts for drainage of amniotic fluid in cases of hydrothorax and FETO in patients with congenital diaphragmatic hernia that met the criteria of being 26 weeks or less of gestational age and with factors associated to high mortality, such as a lung-to-head ratio (LHR) of less than 1 or herniation of the liver into the thoracic cavity.
The diagnosis of PH was based on pulmonary perfusion scintigraphy, chest CT and clinical criteria. The criteria for diagnosis were reduced pulmonary perfusion in scintigraphy and reduced volume of the lung and pulmonary vessels of the involved side compared to the contralateral side in CT. The clinical diagnosis was based on the presence of respiratory distress syndrome of the neonate, the need for mechanical ventilation at high pressures and plain chest radiography showing reduced aeration and small lung fields.
Lung function was assessed in cooperative children aged more than 5 years by means of forced spirometry, plethysmography and single breath diffusion (DLCO) conforming to the standards set by the European Respiratory Society (ERS). Testing was performed with a Jaeger® spirometer and a Jaeger Masterscreen™ plethysmograph (CareFusion, San Diego, USA). We have expressed the data as z-scores adjusted for sex, age and height based on the reference equations of the Global Lung Initiative (GLI) of 2012 and Rosenthal, respectively.
To describe quality of life, we reviewed data from the medical records of yearly check-up visits on factors such as schooling, type of physical activity performed by the child, and limitations in physical activity compared to healthy children of the same age. We analysed survival by means of the Kaplan-Meyer method using SPSS version 21.
ResultsWe identified 60 cases, 33 of which corresponded to male patients (55%). There was unilateral involvement in 35 cases (21 left and 15 right) and bilateral involvement in 24 cases. All patients had been assessed before birth by means of 3D ultrasound, and abnormalities had been found in forty-six: oligohydramnios in nineteen, diaphragmatic hernia in twenty-one, hydrothorax in six, Jeune syndrome in two, diaphragmatic eventration in one, and polyhydramnios in one. The mean gestational age at the first abnormal ultrasound was 27 weeks (range, 15–39 weeks). Foetal MRI was performed in seven patients, confirming the presence of congenital diaphragmatic hernia with reduced size of the ipsilateral lung in six patients and arthrogryposis with reduced size of both lungs in one patient.
PH was classified as primary in four patients and secondary in fifty-six, and congenital diaphragmatic hernia was the most frequent cause of PH (41%). Table 2 presents the aetiologies found in our study. The diagnoses associated with the 19 cases of oligohydramnios were renal or urinary tract malformation (6 cases), premature rupture of membranes (5 cases, 3 of which were due to an invasive prenatal intervention), hydrops fetalis (3 cases) and congenital diaphragmatic hernia (1 case), while the aetiology was not established in one case. The mean LHR in patients with congenital diaphragmatic hernia was 0.89 (range, 0.29–4.6). Foetal surgery was performed in fifteen patients following the detection of abnormalities in ultrasound, FETO was performed in eleven cases, and a pleural-amniotic shunt placed in five.
Distribution of aetiologies and outcomes of cases of pulmonary hypoplasia diagnosed between 1995 and 2014.
| Causes of pulmonary hypoplasia | Number of cases (%) | Deceased patients | Patients that required mechanical ventilation in the neonatal period | Patients that received home mechanical ventilation |
|---|---|---|---|---|
| Diaphragmatic hernia | 24 (40) | 9 | 23 | 1 |
| Left | 16 | |||
| Bilateral | 3 | |||
| Right | 5 | |||
| Diaphragmatic paralysis-eventration | 2 (3,3) | 1 | 1 | |
| Renal/urinary tract malformation | 6 (10) | 2 | 6 | 3 |
| Invasive prenatal intervention | 3 (5) | 3 | 3 | |
| Hydrops fetalis | 3 (5) | 3 | 3 | |
| Premature rupture of membranes | 2 (3,3) | 2 | 2 | |
| Oligohydramnios of unknown aetiology | 2 (3,3) | 1 | ||
| Congenital hydrothorax | 2 (3,3) | 1 | 1 | |
| Jeune syndrome | 2 (3,3) | 1 | 1 | 1 |
| Scimitar syndrome | 5 (8,3) | |||
| Intrathoracic masses | 2 (3,3) | 1 | 1 | |
| Unknown aetiology (associated with Down syndrome) | 1 (1,6) | 1 | 1 | |
| Foetal akinesia/hypokinesia sequence | 2 (3,3) | 2 | 2 | |
| Primary pulmonary hypoplasia | 4 (6,6) | 2 | 1 | 1 |
The mean gestational age at birth was 33 weeks (range, 27–39 weeks). Fifty patients had respiratory distress at birth, of whom forty-seven (94%) required mechanical ventilation due to severe hypoxaemia for a mean of nine days. Eight patients had persistent pulmonary hypertension of the newborn.
The onset of symptoms occurred later in six patients that received a diagnosis of PH after being assessed for recurrent episodes of bronchitis. The mean chronological age of diagnosis of these six patients was 45 days (range, 30–180). Four patients did not develop any clinically significant respiratory symptoms and received the diagnosis of PH following the detection of dextrocardia in the first year of life, and the presence of scimitar syndrome was subsequently confirmed in all.
The diagnosis of PH was suspected in 18 cases of early neonatal death (31%) based on clinical features and compatible plain radiography findings. In 31 cases (51.6%) PH was diagnosed based on chest CT or lung perfusion scintigraphy: both techniques in 14 cases, CT alone in 11 cases, and scintigraphy alone in 6 cases. In patients that underwent scintigraphy, the average perfusion in the affected lung was 29% (range, 8–44%). The diagnosis was confirmed by autopsy in 11 patients (19%).
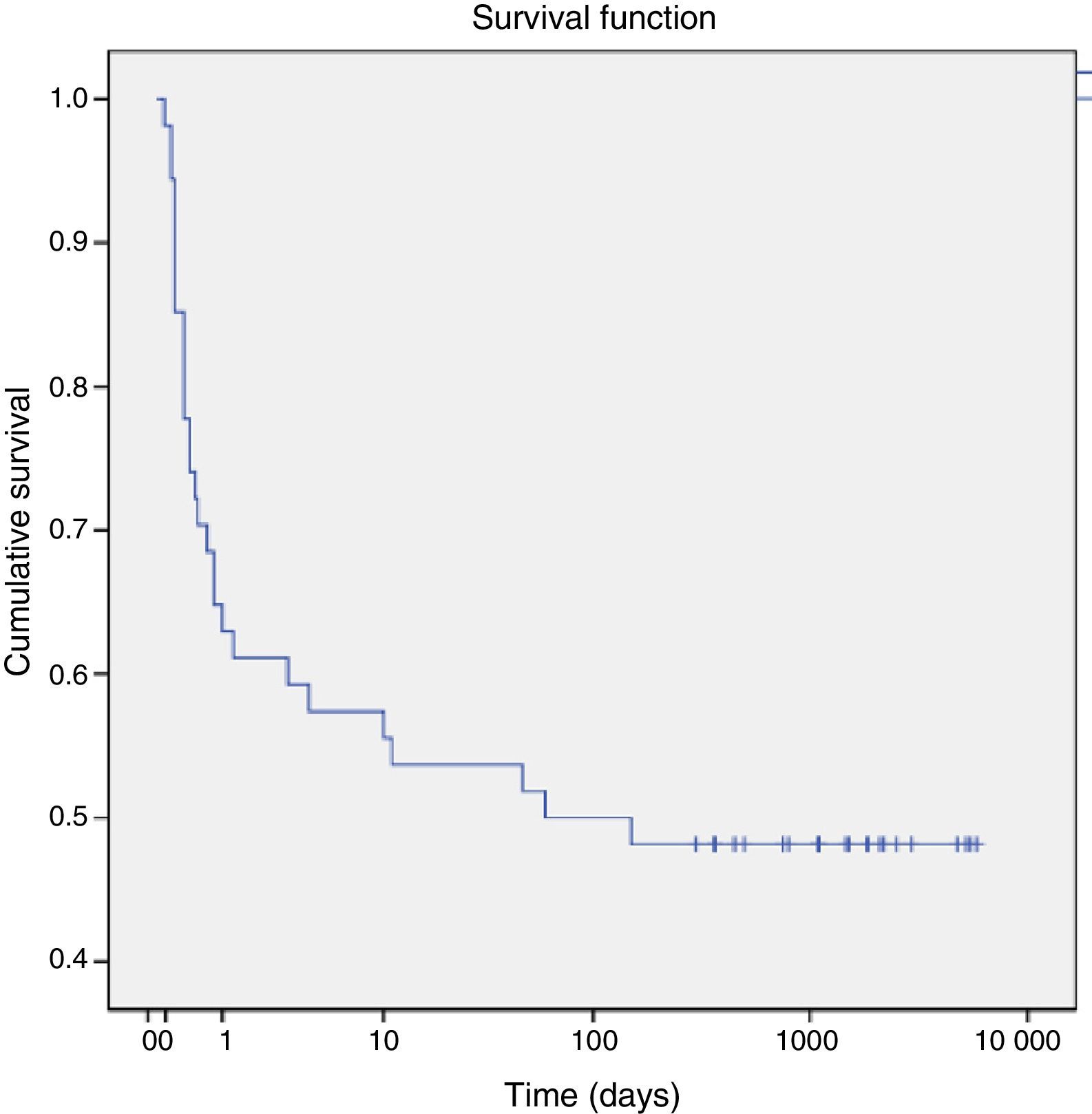
As for mortality, a total of 28 patients (47%) died in the first 60 days post birth, with 25 dying in the perinatal period. Seventy-five percent of the deaths occurred in the first day post birth. Using the Kaplan–Meier method, we estimated that the cumulative probability of death was 25% at 12h post birth and increased to 50% by 60 days post birth (Fig. 1).
Four patients were lost to followup, while another 28 were followed up for an average of 5.8 years (range, 0.9–16). Of all the patients that had required ventilatory support in the neonatal period, six went on to require home mechanical ventilation (two of them via tracheostomy) for a median of 36 months, and five required home oxygen therapy for a median of 4 months (range, 2–10 months). One patient developed pulmonary hypertension. Seventeen patients had recurrent respiratory conditions that were classified as pneumonia and recurrent bronchitis, and four developed scoliosis. We analysed the lung function tests performed in the past five years (Table 3). Spirometry manoeuvres met the acceptability criteria in 13 out of a total of 28 patients (42.8%; age range, 5–17 years), and plethysmography results were technically adequate in 11 of these 13 children. Five patients had an obstructive pattern (FEV1/FVC<lower limits of normal [LLN]); two had a restrictive pattern (FVC<LLN and FEV1/FVC>LLN and TLC>LLN); and six had a normal pattern, of whom two had air trapping. Three patients had increased airway resistance. Lung diffusion tests were normal in all 11 patients.
Results of the analysis of lung function tests performed in the past five years in children with pulmonary hypoplasia diagnosed between 1995 and 2014 at the Hospital Universitario Vall d’Hebron de Barcelona.
| Patient | FVC z-scorea | FEV1 z-scorea | FEV1/FVC z-score | TLC z-scoreb | Interpretation |
|---|---|---|---|---|---|
| 1 | –2.06 | –2.7 | –1.55 | – | Obstructive |
| 2 | –2.85 | –3.36 | –1.70 | –0.59 | Obstructive |
| 3 | 1.18 | –0.37 | –2.13 | – | Normal |
| 4 | –2.29 | –2.41 | –0.44 | –1.88 | Restrictive |
| 5 | –2.67 | –3.43 | –1.90 | –0.69 | Obstructive |
| 6 | –0.19 | 0.08 | 0.37 | 0.05 | Normal |
| 7 | –2.12 | –3.92 | –3.12 | –0.25 | Obstructive |
| 8 | –2.84 | –2.7 | 0.15 | –2.72 | Restrictive |
| 9 | –0.49 | –1.25 | –1.35 | 1.96 | Normal |
| 10 | –0.9 | –1.01 | –0.37 | –0.08 | Normal |
| 11 | 0.67 | –1.14 | –1.00 | 0.14 | Normal |
| 12 | –2.48 | –2.24 | 0.35 | –0.31 | Obstructive |
| 13 | 0.73 | 0.84 | 0.00 | 0.42 | Normal |
| Median z-score (range) | –2.06 (–2.84 to 1.18) | –2.55 (–3.92 to 0.84) | –1 (–3.12 to 0.37) | –0.25 (–1.88 to 1.96) |
Twenty-two patients were able to attend school normally and had a normal exercise tolerance for their age.
DiscussionPulmonary hypoplasia results from an arrest in normal lung development due to various factors that are not yet fully understood. We present a detailed review of the patients that received a PH diagnosis in the past 20 years and were managed by an interdisciplinary team at a tertiary care university hospital. We analysed its various causes and clinical presentation, methods used for diagnosis, treatments and outcomes.
The aetiological distribution of the cases described in this study is consistent with the data found in the literature,8,20,21 according to which the most common congenital anomalies associated with PH are diaphragmatic hernia, renal anomalies and skeletal anomalies. We ought to mention other diseases found in our review, such as premature rupture of membranes caused by invasive prenatal procedures such as multifoetal pregnancy reduction or amniocentesis, genetic disorders (trisomy 21), and some that are less understood, such as foetal akinesia/hypokinesia sequence or Pena-Shokeir syndrome (2 cases in our series), characterised by polyhydramnios, intrauterine growth restriction, craniofacial and limb anomalies, arthrogryposis, short umbilical cord, PH, and a very low survival.22,23 Cases of primary PH are rare and can be unilateral or lobar.3 In our series, only 7.8% of the cases were classified as primary PH.
Oligohydramnios was the mechanism associated with PH in one third of the cases in our series. Nimrod et al.18 described a greater impact of premature membrane rupture in foetuses in which rupture occurred before 26 weeks’ gestation and lasted more than five weeks, while Rotschild et al.20 detected pulmonary hypoplasia in up to 14% of foetuses that experienced oligohydramnios following the rupture of membranes before 29 weeks’ gestation.
Other authors have analysed the correlation between gestational age and the risk of having PH in association with oligohydramnios, concluding that a duration of oligohydramnios of more than 14 days following the rupture of membranes before 25 weeks’ gestation carries a risk of death due to PH of up to 90%.21,22 While we did not establish patient subsets in our study to analyse the impact of oligohydramnios based on its duration and gestational age at the time of diagnosis, we found that the mean gestational age at the time oligohydramnios was detected was 27 weeks, which is consistent with the literature.
Also consistently with the literature, the congenital malformation most frequently associated with PH was diaphragmatic hernia.10,24,25 This disease is the model that has been used most commonly for the study of early interventions, the pathophysiology, advances in perinatal care and the development of new diagnostic tools in PH.26 A distinctive aspect of congenital diaphragmatic hernia is that PH develops as an inherent feature of the disease and not solely as a result of lung compression, and the mechanism by which the lung contralateral to the hernia is also affected remains unknown.17,18
The prenatal diagnosis of PH and its confirmation in the early neonatal period are key in decreasing the morbidity and mortality associated to this disease. Consequently, the routine use of 3D ultrasound and MRI for prenatal diagnosis has become a care standard.13,16,27,28 The current ERS guidelines for the management of congenital diaphragmatic hernia include the use of MRI for the prenatal assessment of affected foetuses, as it performs better than ultrasound in detecting herniation of the liver into the thorax, differentiating diaphragmatic eventrations and accurate volumetry of both the contralateral and ipsilateral lungs.29 Levine et al.27 performed MRI scans on 74 foetuses in whom ultrasound had detected thoracic abnormalities, confirming that the use of a complementary noninvasive technique contributed additional information in up to 38% of cases and led to changes in the management of 8% of them. All our patients were assessed by prenatal 3D ultrasound, and patients with severe diaphragmatic hernia were also assessed by MRI.
Of the 24 patients in whom congenital diaphragmatic hernia was diagnosed as the cause of PH, 11 were classified as being at high risk of dying and met the criteria for prenatal surgery, so they underwent FETO. Invasive prenatal surgical procedures have evolved from open surgery with placement of a tracheal clip to the FETO technique in which, after ascertaining lung growth, the balloon is removed prenatally. These techniques have led to an increase in postnatal survival and a decrease in the incidence of secondary pulmonary hypertension.15,30,31
After birth, PH may be suspected when plain radiography findings include diaphragmatic domes elevated up to the seventh rib, a bell-shaped thorax, downward sloping ribs, pneumothorax or pneumomediastinum.2,3,32 Pulmonary perfusion scintigraphy and chest computed tomography are very useful in confirming the diagnosis.5,32–37 In our series, 50.4% of the cases were diagnosed by scintigraphy and CT, 31% by plain radiography and compatible clinical features (cases that could not be confirmed by means of other techniques due to the early death of the patients), and 19% by autopsy. The average measurement of perfusion by scintigraphy is used to determine the severity of PH, which is considered moderate if perfusion in the involved lung is less than 40%.33–35
The perinatal mortality observed in our series was 41.6%, which was consistent with the various series published in the literature that have described a high perinatal mortality.2,4,10,15,25 When we analysed our data, we found that the cumulative probability of dying in the first 12h post birth was 25%, and that this probability rose to 50% by 60 days post birth, a finding that underscored the need to have a highly specialised interdisciplinary team for the management of these patients during the early neonatal period. The clinical manifestations observed most frequently during followup were pneumonia and recurrent bronchitis, consistent with the course of disease described in patients that have undergone surgical repair of congenital diaphragmatic hernia and cases of late onset with similar symptoms.29,37 Twenty-five percent of the patients that required mechanical ventilation in the neonatal period and survived required home mechanical ventilation and oxygen supplementation for a median of four months. These data differ from the current literature, which describes a shorter duration of respiratory support.34 This could be due to a lack of descriptive studies similar to the one we have performed in our population. On the other hand, 16% of our patients developed scoliosis secondary to the unilateral reduction in lung volume. Previous studies have shown that up to 16% of children with a previous history of diaphragmatic hernia have some type of thoracic asymmetry, such as scoliosis or pectus carinatum.34 Other longitudinal studies in children that had undergone surgical repair of diaphragmatic hernia showed catch-up lung growth and adequate exercise tolerance, with children being able to lead normal lives despite the persistence of asymmetry in radiological images and a lack of changes in perfusion scans.19 Our study showed similar results, as 77% of the patients that survived in our series could attend school normally and had an adequate exercise tolerance for their age.
When it comes to lung function tests, some authors have described an obstructive airway pattern in patients with HP and proposed that bronchoscopy be performed to rule out tracheobronchomalacia.38 Most studies have reported a predominance of restrictive airway patterns, especially in patients requiring prolonged mechanical ventilation or oxygen therapy.39,40 In our case series, few data on lung function were available because we only took into account the data collected in the past five years, during which lung function testing was performed by the same technicians with the same equipment and adhering to the stringent quality and acceptability criteria published by the ERS, to avoid potential biases. Furthermore, most of the patients in followup could not cooperate with spirometry or plethysmography. Thus far, our results have shown a higher proportion of children in followup with an obstructive pattern compared to a restrictive pattern based on spirometry as well as plethysmography.
In the past two decades, there have been advances in the diagnostic techniques and prenatal interventions for PH, and postnatal care for this disease has been optimised. This has led to the improved management of these patients, although the associated morbidity and mortality continue to be considerable. We believe that further studies are required to assess the impact of these early interventions on future lung development. Furthermore, we must continue to do research to identify other factors that would allow changing the course of PH earlier and by less invasive methods.
ConclusionsPH is a congenital anomaly that can be due to various causes and the prevalence of which is underestimated. It is associated with a high neonatal mortality and significant long-term morbidity, which calls for early diagnosis and intervention by a specialised interdisciplinary team. The development of new diagnostic and surgical techniques has improved survival in these patients, but further research is necessary to identify new factors that would allow changing the course of this disease, along with long-term studies to assess the impact of these interventions.
Conflict of interestsThe authors have no conflict of interests to declare.
Please cite this article as: Delgado-Peña YP, Torrent-Vernetta A, Sacoto G, de Mir-Messa I, Rovira-Amigo S, Gartner S, et al. Hipoplasia pulmonar: análisis de la casuística durante 20 años. An Pediatr (Barc). 2016;85:70–76.
