




Tuberous sclerosis (TS) is an inherited disorder with multisystemic involvement and a high phenotypic variability. There are two genes that cause this condition: TSC1 and TSC2.
ObjectivesOur goal was to clinically characterise patients with TS followed up in the Pediatric Neurology Clinic of a tertiary hospital during the last 10 years, and correlate the genotype with the severity of neurological manifestations and imaging studies.
Patients and methodsRetrospective analysis of patients with TS, including review of medical records and available MRI imaging.
ResultsWe studied 35 cases with a median age at diagnosis of 10 months. Seizures were the first manifestation in 91.4% of cases, with a predominance of epileptic spasms. Over 50% had cognitive impairment and 49% behavioural disorders.
A genetic study was performed on 24 children, and TSC2 mutations identified in 58.3% of them. Of the 11 cases of refractory epilepsy, six had the TSC2 gene mutation. In the group of eight patients with moderate/severe cognitive deficits, five had TSC2 mutations.
We reviewed 26 MRI scans, in which it was observed that 76.9% had diffuse involvement of cerebral lobes, which reflects a greater burden of injury. Of the patients who had an MRI scan performed and had TSC2 mutations, all had a high tuber load, and 5 of them had refractory epilepsy.
DiscussionIn our sample we observe a high percentage of mutations in the TSC2 gene. This mutation carries a worse neurological prognosis, with drug-resistant epilepsy and a more severe cognitive impairment.
La esclerosis tuberosa (ET) es una enfermedad de afectación multisistémica y gran variabilidad fenotípica. Están identificados 2 genes involved en la génesis de la enfermedad: TSC1 y TSC2.
ObjetivosCaracterizar clínicamente a los pacientes con ET seguidos en Neurología Pediátrica de un hospital de tercer nivel durante los últimos 10 años y correlacionar el genotipo con la gravedad de la clínica neurológica y los estudios de imagen.
Pacientes y métodosEstudio retrospectivo descriptivo, mediante consulta de la historia clínica y evaluación de las resonancias magnéticas (RM) de pacientes con ET.
ResultadosSe estudiaron 35 casos, con una mediana de edad, al diagnóstico, de 10 meses. En el 91,4% se registraron crisis epilépticas, con un predominio de espasmos epilépticos a la presentación.
Más del 50% tenía deterioro cognitivo y el 49% trastornos de conducta.
Se sometieron a estudio genético 24 niños con predominio de mutaciones TSC2 (58,3%). De los 11 casos de epilepsia refractaria, 6 tenían mutación del gen TSC2. De los 8 pacientes con déficit cognitivo moderado a grave, se identificaron 5 mutaciones TSC2.
Se revisaron 26 RM y en el 76,9% se observó una afectación completa de los lóbulos cerebrales, lo que refleja una gran cantidad de lesiones. De los enfermos con mutaciones TSC2 y RM realizada, todos tenían alta carga de lesión y 5 epilepsia refractaria.
DiscusiónEn nuestra muestra, nos encontramos con un alto porcentaje de mutaciones en el gen TSC2. Esta mutación está asociada a un peor pronóstico neurológico, con crisis más farmacorresistentes y un atraso cognitivo más severo.
Tuberous sclerosis (TS) is a multisystem neurocutaneous disorder. Its observed features are the result of disrupted cell differentiation, proliferation, and migration in the early stages of foetal development.1
Tuberous sclerosis is an autosomal dominant genetic disorder with an incidence of approximately 1 in 5000–10,000 births.2 So far, 2 genes have been identified that are involved in its aetiology: TSC1 (chromosome 9q34) and TSC2 (chromosome 16p13.3). Only 7–37% of patients have a positive family history of TS, as most cases (65–75%) correspond to de novo mutations.3 There was also a highly variable range of phenotypic expression, age at onset, signs and symptoms, and severity of disease.
A disease-causing mutation is found in 60–89% of patients that meet the criteria for TS, and it is estimated that approximately 50% correspond to mutations in the TSC2 gene and 17% to mutations in the TSC1 gene.4–6
TS is characterised by the development of benign tumours in multiple organs.7 In addition, there is an increased risk of malignancy.8
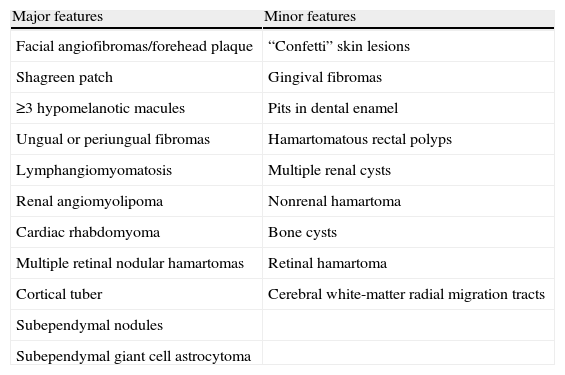
The diagnosis of TS is based solely on clinical criteria (Table 1). It is categorised as definite TS if 2 major features or 1 major feature and 2 minor features are present; as probable TS if 1 major feature and 1 minor feature are present; and possible TS if 1 major feature or 2 or more minor features and no major features are present. Genetic testing is useful for family studies or to confirm TS.
Diagnostic criteria for tuberous sclerosis: Roach.
| Major features | Minor features |
| Facial angiofibromas/forehead plaque | “Confetti” skin lesions |
| Shagreen patch | Gingival fibromas |
| ≥3 hypomelanotic macules | Pits in dental enamel |
| Ungual or periungual fibromas | Hamartomatous rectal polyps |
| Lymphangiomyomatosis | Multiple renal cysts |
| Renal angiomyolipoma | Nonrenal hamartoma |
| Cardiac rhabdomyoma | Bone cysts |
| Multiple retinal nodular hamartomas | Retinal hamartoma |
| Cortical tuber | Cerebral white-matter radial migration tracts |
| Subependymal nodules | |
| Subependymal giant cell astrocytoma |
Neurologic symptoms are present in 85% of cases and constitute the main cause of morbidity and mortality.3 Epilepsy and cognitive deficits are commonly associated to brain lesions, including glioneuronal hamartomas (also called tubers), white matter lesions, and subependymal giant cell astrocytomas.9
Epilepsy usually appears in the first year of life, and infantile spasms are a common presentation (36–96%).10 The number of glioneuronal hamartomas, and especially the proportion of the total brain volume they occupy, are associated with the presence of severe cerebral dysfunction (refractory epilepsy and/or moderate to severe cognitive impairment).1,11 The association between the severity of seizures and the presence of cyst-like cortical tubers has also been established.12
It is estimated that 50% of TS patients have cognitive impairments, the severity of which is associated with having a history of infantile spasms or refractory epilepsy, and to the number and proportion of brain volume occupied by glioneural hamartomas.13
Behavioural problems are present in 40–90% of TS patients. Although behavioural changes may develop irrespective of the level of cognitive function, it has been demonstrated that cognitive disability and greater seizure frequency are risk factors for the presence of behavioural difficulties.14
Dermatologic features can appear in 81–95% of TS cases, the most common being hypomelanotic macules, angiofibromas, ungual fibromas, Shagreen patches, and fibrous plaques.10,15 There is no significant risk of malignant transformation of skin lesions.
Between 50% and 60% of TS patients have cardiac manifestations, with rhabdomyoma being the most frequent. They are benign tumours, often multifocal, and usually asymptomatic. Most rhabdomyomas regress spontaneously.
Renal features are found in 60–80% of TS patients, and the most common form is angiomyolipoma. Renal lesions are usually benign and carry a haemorrhage risk in relation to their size. They also carry a high risk of developing renin-dependent hypertension and chronic renal disease due to the compression and replacement of renal parenchyma. Renal cell carcinoma develops in approximately 1–2% of adults, so regular monitoring with ultrasonography is recommended.
Ophthalmic manifestations are present in 87% of TS patients. Although they are useful in making the diagnosis, they rarely affect visual acuity, and do not require specific treatment.16
Genotype-phenotype correlations in TS have been investigated since the 2 genes associated with the disease were first identified. Several studies have concluded that mutations in the TSC1 gene are associated to a milder phenotype than TSC2 mutations, but there is disagreement on this point.15–17 While mutations in the TSC2 gene are usually associated with more severe phenotypes (with increased frequency of refractory epilepsy, cognitive impairment, and behavioural changes), mild phenotypes have also been described for TSC2 mutations.17
ObjectivesOur aim was to analyse the clinical characteristics of TS patients followed in the paediatric neurology department of the Centro Hospitalario del Porto (CHP) in the past 10 years, and to correlate patient genotypes with the severity of neurologic manifestations and radiographic findings.
Patients and methodsWe conducted a descriptive retrospective analysis of patients with definitive TS (based on the clinical diagnostic criteria) followed in the paediatric neurology department of a tertiary hospital, the CHP, in Porto, Portugal. Data were collected from electronic medical histories and paper clinical notes.
We used Microsoft Office Excel 2007 to perform the descriptive statistical analysis. The variables analysed were: current age; sex; age at diagnosis; family history; presence and presentation of epilepsy; psychomotor development; behavioural, cutaneous, cardiac, renal, and ophthalmic abnormalities; and genetic study.
All the magnetic resonance (MR) images analysed were reviewed independently by a single neuroradiology specialist, and used to investigate the following variables: number of tubers, supratentorial or infratentorial location, number of cerebral lobes with lesions, number and lateralisation of cyst-like tubers, radial migration lines in the white matter, subependymal nodules, and giant cell astrocytomas. MR imaging was performed on a 1.5T GE Signa system or on a 3.0T TX Philips Achieva system (3 patients) in the neuroradiology department of the CHP. A preliminary localising scan in the sagittal plane was used to identify anterior and posterior commissures and to position the axial images in parallel to the intercommissural plane: FLAIR, T2 SE, T2 *, T1 SE and T1 SE post-contrast.
FLAIR images were used to identify tubers and radial migration lines,18 and T2-weighted FLAIR images to identify subependymal nodules.
Tubers were defined as corticosubcortical lesions with high signal intensity on FLAIR images, and radial migration lines as lines of hyperintensity on FLAIR images extending from the ventricles to tubers in the cortex. Subependymal nodules were found in all ventricles, protruding into the ventricles from the ventricular wall.
Tubers with subcortical cysts were defined as lesions with cerebrospinal fluid signal characteristics on MRI bordered by a hyperintense rim in T2 scans. Tubers and Calcified tubers and subependymal nodules showed low signal in T2 and T2* scans. Since the density of tubers in brain parenchyma is suggested as the best marker of severity of epilepsy and cognitive function1, we performed a qualitative assessment of brain tubers density by estimating the frequency of tubers in each cerebral lobe. The density of tubers reflects brain lesion load, obtained by evaluation of the total number of tubers and involvement of each brain lobe by tubers. High brain lesion load was considered if all cerebral lobes contained tubers in patients with total number of more than 15. Quantitative assessment with automatic segmentation of tuber density or brain volume occupied by tubers was not performed due to inaccessibility to appropriate software.
Formal cognitive assessments were performed depending on the age group with the Ruth Griffiths mental development scale and/or the Wechsler scale (WISC-R or WISC-III) in the psychology clinic of the CHP. Cognitive deficit was defined as an intellectual quotient (IQ) below 70, with the following severity levels: mild, with IQ ranging from 50–55 to 70; moderate ranging from 35–40 to 50–55; severe 20–25 to 35-40; and profound, less than 20–25. Cognitive deficit was classified as “unspecified” when it had not been assessed but it was presumed (with IQ <70).19
The genetic studies were performed in 2 specialised laboratories: the Centro de Genética Médica (Centre of Medical Genetics), Porto, through the Erasmus MC University Medical Center in Rotterdam, using direct sequence analysis and multiplex ligation-dependent probe amplification analysis; and in the Centro de Genética Clínica (Centre of Clinical Genetics), Porto, by polymerase chain reaction (PCR) and direct sequencing of the full encoding region of the TSC1 and TSC2 genes and the adjacent intron regions; quantitative PCR was also performed to study deletions of the TSC2 gene.
Refractory epilepsy was defined as failure to control seizures despite treatment with 2 or more appropriately chosen and administered antiepileptic drugs.20
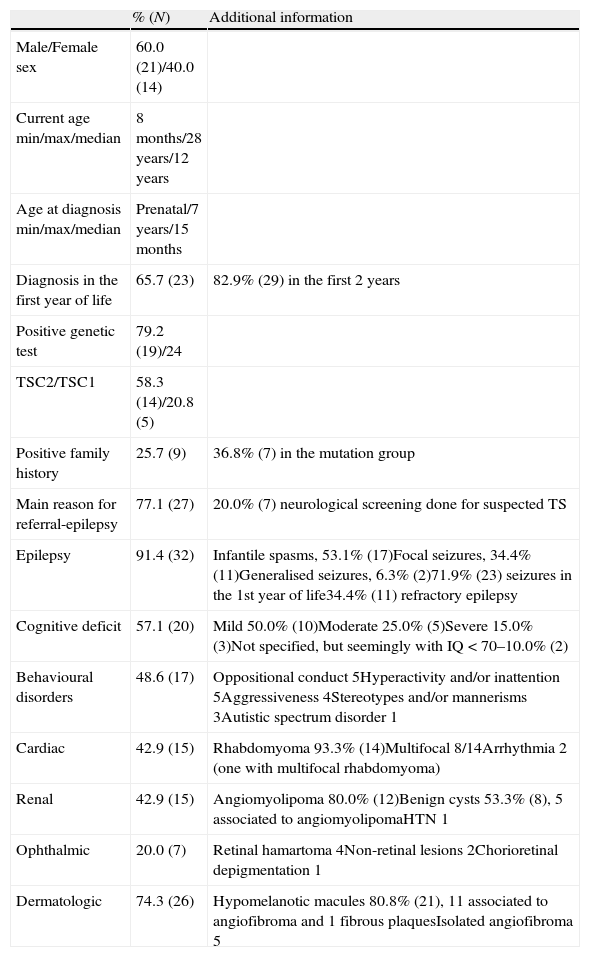
ResultsWe analysed 35 cases of definitive TS, characterised in Table 2.
Clinical characteristics of 35 patients with tuberous sclerosis.
| % (N) | Additional information | |
| Male/Female sex | 60.0 (21)/40.0 (14) | |
| Current age min/max/median | 8 months/28 years/12 years | |
| Age at diagnosis min/max/median | Prenatal/7 years/15 months | |
| Diagnosis in the first year of life | 65.7 (23) | 82.9% (29) in the first 2 years |
| Positive genetic test | 79.2 (19)/24 | |
| TSC2/TSC1 | 58.3 (14)/20.8 (5) | |
| Positive family history | 25.7 (9) | 36.8% (7) in the mutation group |
| Main reason for referral-epilepsy | 77.1 (27) | 20.0% (7) neurological screening done for suspected TS |
| Epilepsy | 91.4 (32) | Infantile spasms, 53.1% (17)Focal seizures, 34.4% (11)Generalised seizures, 6.3% (2)71.9% (23) seizures in the 1st year of life34.4% (11) refractory epilepsy |
| Cognitive deficit | 57.1 (20) | Mild 50.0% (10)Moderate 25.0% (5)Severe 15.0% (3)Not specified, but seemingly with IQ<70–10.0% (2) |
| Behavioural disorders | 48.6 (17) | Oppositional conduct 5Hyperactivity and/or inattention 5Aggressiveness 4Stereotypes and/or mannerisms 3Autistic spectrum disorder 1 |
| Cardiac | 42.9 (15) | Rhabdomyoma 93.3% (14)Multifocal 8/14Arrhythmia 2 (one with multifocal rhabdomyoma) |
| Renal | 42.9 (15) | Angiomyolipoma 80.0% (12)Benign cysts 53.3% (8), 5 associated to angiomyolipomaHTN 1 |
| Ophthalmic | 20.0 (7) | Retinal hamartoma 4Non-retinal lesions 2Chorioretinal depigmentation 1 |
| Dermatologic | 74.3 (26) | Hypomelanotic macules 80.8% (21), 11 associated to angiofibroma and 1 fibrous plaquesIsolated angiofibroma 5 |
HTN: Hypertension.
A genetic study was done in 24 children, and a mutation identified in 79.2% (19) of the cases, with TSC2 mutations being most frequent (58.3%) compared to TSC1 mutations (20.8%).
There was a positive family history in 25.7% (9) of the cases: a single first-degree family member was affected in 3 cases; 2 family members were affected in 2 cases, and in 4 cases, first-degree family members (parents or siblings) were diagnosed after the patient's own diagnosis.
In the group with identified mutations, 36.8% of patients had a positive family history: in 40.0% of patients with a TSC1 mutation (2/5) and 35.7% (5/14) of patients with a TSC2 mutation.
The reasons for referral to Paediatric Neurology consultation were: epilepsy in 77.1% (27) of cases, delay in psychomotor development associated to MRI abnormalities in 1 case, and neurologic disease screening in children with suspected TS in 7 cases (6 cases of cardiac hamartoma, 2 of which were associated to skin abnormalities; and 1 with a positive family history and skin abnormalities).
Epileptic seizures occurred in 91.4% (32) of the patients. Infantile spasms were the most frequent presentation, found in 53.1% (17). In 71.9% (23) of cases, the seizures started in the first year of life, and 34.4% (11) of the patients had refractory epilepsy.
Of the patients with well-controlled seizures, 11 were currently taking an antiepileptic drug, 2 were taking other medication, and 7 were not taking any medication. Only 1 case was treated with surgery and had a favourable outcome. Vigabatrin was the drug of choice in the treatment of infantile spasm (88.2%) as monotherapy or polytherapy.
Cognitive impairment was documented in 57.1% (20) of children, and behavioural disorders in 48.6% (17).
Cardiac manifestations were present in 42.9% (15) of patients, renal manifestations in 42.9% (15), and ophthalmic manifestations in 20.0% (7).
Dermatologic features were identified in 74.3% (26) of patients, and malignant transformations were not observed in any of the cases.
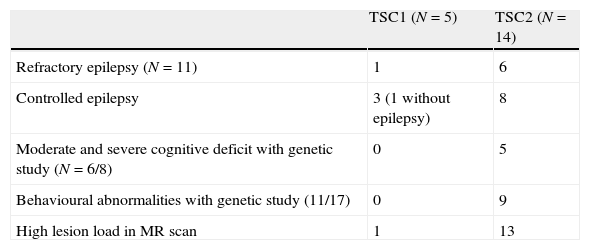
When we correlated the genotype to the clinical phenotype (Table 3), 6 of the 11 cases of refractory epilepsy were associated with mutations in TSC2 and only 1 case to a mutation in TSC1.
Of the remaining patients with TSC1 mutations, 1 had no seizures and 3 had well-controlled epilepsy. Among the patients with TSC2 mutations, 8 had well-controlled epilepsy (6 with pharmacological treatment, and 2 without).
A genetic study was performed in 6 of the 8 patients with moderate and severe cognitive deficit. None of these patients had a TSC1 mutation, and TSC2 mutations were identified in 5. Genetic studies were also performed in 11 patients with behavioural difficulties, identifying TSC2 mutations in 9 of the patients, and no TSC1 mutations.
Of the 8 patients with moderate and severe cognitive impairment, 5 had onset of seizures before 12 months of life. The 3 cases of severe cognitive delay were associated with refractory epilepsy and behavioural difficulties. However, 3 of the 16 cases of early onset epilepsy and infantile spasms had a normal psychomotor development (formal cognitive assessment performed at 3, 7, and 11 years of age).
In the 74.3% (26) patients for whom MRIs were available, 76.9% (20) of them had tubers in every cerebral lobe and more than 15 tubers in total, thus presenting high lesion load. Radial migration lines were observed in 96.2% (25) of the patients (Fig. 1). Subependymal nodules were found in 84.6% (22), most of them calcified, and 3 of them enhancing (Fig. 2). Infratentorial tubers were found in 23.1% (6) of the patients. Cyst-like tubers were present in 7 (26.9%) patients, with juxtacortical location in 6, and infratentorial location in 1 (Fig. 3). There was one probable case of giant cell astrocytoma, around the foramen of Monro, measuring less than 1cm, without hydrocephalus (Fig. 4). Focal atrophy was not found in any of the patients.
 and radial migration lines.")



We reviewed the MR studies of all patients with an identified TCS2 mutation except one, and found a high lesion load in all of them. One of the 5 patients with a TSC1 mutation had a high lesion load. Of the 5 cases associated with refractory epilepsy, 2 had cyst-like tubers.
We found high lesion loads in 87.5% of the patients with cognitive delays and in 78% of patients in whom seizures appeared before 12 months of age. We also found radial migration lines in 89% of the children with early onset of epilepsy.
DiscussionTuberous sclerosis is a multisystem disease with variable phenotypic expression, and its clinical manifestations may be subtle, nonspecific, and have onset at different ages, so diagnosis is often delayed.21 Diagnosis occurred early in our series, perhaps because the main reason for referral to the department was infantile spasms, a condition whose association with TS is well known by health care professionals, may have facilitated the diagnosis.
Mutations were identified in 79.2% of the children who were subject to genetic studies, with predominance of TSC2 mutations, consistent with other publications.4–6
The number of cases with a positive family history was small,3 and it was greater in the group of patients with identified mutations. In 4 cases, the family history was only identified in first-degree relatives (parents or siblings) after the patient was diagnosed, underscoring the importance of assessing the parents and siblings of children diagnosed with TS without a positive family history for the disease.22
As mentioned above, epilepsy was the main reason for referral,1 manifested in most cases as infantile spasms with onset in the first year of life.
Disease severity and refractory epilepsy were positively correlated to early onset of seizures: seizures started before 12 months of age in all cases with refractory epilepsy.
Vigabratin was the drug used most often to treat infantile spasms in our TS patients (88.2%), consistent with the recommendations of researchers at the National Institute of Health Tuberous Sclerosis Complex Consensus Conference (2000), who considered it the most efficacious treatment option for infantile spasms due to TS.23
Cognitive deficits were documented in a high percentage of cases with great variability in severity, consistent with the literature (44–65%), and demonstrating the importance of making a formal cognitive evaluation in every patient newly diagnosed with TS. The purpose would be to implement early intervention programmes for affected children, and to improve their prognosis by means of cognitive interventions and stimulation.
Our results confirm the positive correlation between psychomotor development delay (PMDD) and epilepsy, as we observed onset of epilepsy in the first year of life and progression to refractory epilepsy in all cases with severe cognitive impairment.9 On the other hand, there were patients in our sample who had infantile spasms and refractory epilepsy without PMDD. Behavioural disorders were very common and were associated with cognitive impairment and refractory epilepsy.
Consistent with previous studies, the most common cardiac manifestation of TS in our sample was rhabdomyoma (14/15), which was multifocal in a majority of cases (8/14). The percentage of renal and ophthalmic involvement was lower than that described in the literature, and no significant complications were documented (there was only one case of hypertension), probably because all our patients were in the paediatric age group. The percentage of cutaneous involvement was lower than what is typically described, which may be due to the poor documentation of these manifestations in the medical records.
Our data supported that neurologic outcomes are worse in patients with TSC2 mutations, showing a greater percentage of drug-resistant epilepsy, more severe cognitive deficits, a higher prevalence of behavioural disorders, and higher lesion loads in MRI.
We found a high lesion load in 87.5% of the patients with cognitive deficits and in 78% of the patients with early-onset seizures, consistent with the association between epilepsy, cognitive function, and lesion load found by other studies.1
In patients that underwent MR imaging, radial migration lines were the most common radiographic alteration (96.2%), approximating the 100% recently described by Van Eeghen et al.24 This group of researchers also reported an association between radial migration lines and early onset of seizures, which was also consistent with our data.
We studied a paediatric sample to increase our knowledge of the main manifestations and comorbidities, the prognosis, and the severity of TS disease in this age group, and to help improve our clinical approach.
There are limitations to this study. It is a retrospective study based on medical records, and it has been conducted in a tertiary centre, with a selective bias for severe cases. We must also note the heterogeneity in the initial clinical evaluation and subsequent follow-up between different specialists, as well as in sample size and composition, which interferes with the statistical significance of our conclusions.
Performance of multicentre studies in the future will help develop a standard approach to the follow-up of these cases.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Monteiro T, Garrido C, Pina S, Chorão R, Carrilho I, Figueiroa S, et al. Esclerosis tuberosa: caracterización clínica e intento de correlación fenotipo/genotipo. An Pediatr (Barc). 2014;81:289–296.
