




La anemia hemolítica autoinmune (AHAI) es una enfermedad rara en niños, generalmente autolimitada.
Material y métodosEstudio descriptivo transversal en menores de 18 años diagnosticados de AHAI desde enero de 1997 a julio de 2019. Se recogieron variables clínicas y se clasificaron según el test de Coombs directo (TCD) en AHAI por anticuerpos calientes (IgG+/-C3d) y fríos (C3d). Se analizó la respuesta al tratamiento y su evolución.
ResultadosSe incluyeron 25 pacientes, siendo el 72% varones. La media de edad al diagnóstico fue de dos años (rango 0,4-9). Los síntomas predominantes fueron fiebre (72%), palidez (68%), ictericia (64%), hepatoesplenomegalia y coluria (48%). La mediana de hemoglobina al diagnóstico fue 5,4 g/dL. En el 96% el TCD fue positivo, con detección de anticuerpos IgG en el 76%. Un solo caso presentó TCD negativo. Un 20% asociaron otra citopenia, uno de ellos fue diagnosticado posteriormente de inmunodeficiencia variable común. En un 32% se sospechó o documentó una infección viral concomitante. La mayoría de los casos fueron autolimitados y respondieron a tratamiento con corticoides (72%). Aquellos con respuesta parcial (24%), principalmente los que asociaban otras citopenias, precisaron otras líneas de tratamiento (rituximab, micofenolato, inmunoglobulinas). Se detectaron complicaciones (32%) y recaídas (26%) únicamente en AHAI por anticuerpos calientes.
ConclusionesNuestra serie confirma que la AHAI es una enfermedad muy poco frecuente en la infancia. La mayoría de los casos evolucionan favorablemente, aunque hasta una cuarta parte precisan segundas líneas de tratamiento y casos excepcionales tratamientos muy agresivos. Estos últimos suelen corresponder a pacientes con más de una citopenia en la evolución de la enfermedad.
Autoimmune hemolytic anemia (AIHA) is a rare and generally self-limiting disease in children.
Material and methodsA descriptive cross-sectional study was performed in children under 18 years diagnosed with AIHA from January/1997 to July/2019. Clinical variables were collected and AIHA was classified according to the direct antiglobulin test (DAT) in warm AIHA (IgG+/-C3d) and cold AIHA (C3d). Response to treatment and evolution were analyzed.
Results25 patients were included and 72% were males. The median age at diagnosis was 2 years (range 0.4 to 9). Fever (72%), pallor (68%), jaundice (64%), hepatosplenomegaly and coluria (48%) were the most common presenting symptoms. The median hemoglobin at diagnosis was 5.4 g/dl. DAT was positive in 96%, with detection of IgG antibodies in 76%. A single case presented negative DAT. 20% of the patients associated another cytopenia, one of which was subsequently diagnosed with common variable immunodeficiency. Concomitant viral infection was suspected or documented in 32%. Most of the cases were self-limiting and responded to corticosteroid treatment (72%). Those with partial response (24%), mainly those associated with other cytopenias, required other lines of treatment (rituximab, mycophenolate, immunoglobulins). Complications (32%) and relapses (26%) were detected only in warm AIHA.
ConclusionsOur case series confirms that AIHA is a very rare disease in childhood. Most cases evolve favorably, although up to a quarter of them require second lines of treatment and, in exceptional cases, they need very aggressive treatments. These latter cases generally correspond to patients who present more than one cytopenia in the course of the disease.
La anemia hemolítica autoinmune (AHAI) es un trastorno inmune caracterizado por la presencia de autoanticuerpos dirigidos contra antígenos de la membrana eritrocitaria que producen acortamiento de la vida media de los hematíes. Es una entidad rara en la infancia, con una incidencia estimada de 0,8-1,25 casos por 100.000 niños1,2, aunque constituye la causa más frecuente de anemia hemolítica extracorpuscular.
Puede aparecer a cualquier edad, con una mayor incidencia en torno a los tres o cuatro años. Etiológicamente, se pueden clasificar en AHAI secundarias y primarias o idiopáticas. En población pediátrica, más de la mitad de los casos se presentan asociadas a otra patología, siendo un 40-50% AHAI idiopáticas3,4.
A diferencia de los adultos, con mayor frecuencia suele presentarse como un cuadro autolimitado asociado a una infección viral. Sin embargo, los menores de dos años y adolescentes presentan más frecuentemente formas crónicas asociadas o no a enfermedades sistémicas, principalmente inmunodeficiencias o trastornos autoinmunes5,6.
La baja prevalencia de este trastorno en población infantil hace que los estudios disponibles sean de carácter retrospectivo, no randomizados y con un escaso tamaño muestral. En consecuencia, existe evidencia científica limitada en el manejo de estos pacientes7.
Este estudio pretende describir las formas de presentación y el diagnóstico de la AHAI en una población pediátrica de un hospital de tercer nivel en los últimos 22 años; así como analizar el tratamiento recibido, la respuesta al mismo y la evolución.
Material y métodosSe realizó un estudio descriptivo transversal en el Hospital Infantil Universitario Niño Jesús, centro de nivel terciario con servicio especializado de Hematología y Oncología Infantil. El período de estudio abarcó desde enero de 1997 hasta julio de 2019.
A través del servicio de codificación, se obtuvo un listado de los pacientes diagnosticados de AHAI durante ese período y se revisaron dichas historias clínicas. Se seleccionaron pacientes menores de 18 años que cumplían la siguiente definición de AHAI: a) presencia de anemia definida como reducción de la concentración de hemoglobina (Hb) por debajo de dos desviaciones estándar de la media de la población general ajustada a la edad; b) evidencia de hemólisis (reticulocitosis, elevación de bilirrubina indirecta, lactato deshidrogenasa [LDH], y/o descenso de haptoglobina); c) estudios inmunohematológicos positivos; d) estudios inmunohematológicos negativos con evidencia de parámetros analíticos de hemólisis, exclusión de causas hereditarias de anemia hemolítica y respuesta al tratamiento estándar.
Las variables recogidas fueron: sexo, edad al diagnóstico, año de debut, presentación clínica, pruebas diagnósticas realizadas, tratamiento recibido y respuesta al mismo, período de seguimiento y la existencia de complicaciones.
Se clasificaron las AHAI según el test de antiglobulina directa o test de Coombs directo (TCD) en: a) producidas por anticuerpos calientes, cuando presentaban IgG positiva con o sin activación de complemento (C3d); b) producidas por anticuerpos fríos cuando únicamente existía activación del complemento (C3d positivo) o c) AHAI TCD negativas.
La respuesta al tratamiento también se clasificó en tres tipos: a) respuesta completa (RC), aquellos que alcanzaban un valor de Hb superior o igual a 11 g/dL junto con cifra de reticulocitos inferior a 120x109/μL permitiendo suspender el tratamiento; b) respuesta parcial (RP), cuando la cifra de Hb se encontraba entre 7-11 g/dL o reticulocitos por encima de 120x109/μL; c) ausencia de respuesta (NR) cuando la Hb era inferior a 7 g/dL o cualquier situación en que es preciso mantener tratamiento de forma sostenida5.
Con los datos obtenidos se realizó un análisis descriptivo y analítico con el programa SPSS v2.0.
ResultadosSe reclutaron 40 pacientes que cumplían los criterios de inclusión. De éstos, 15 pacientes fueron excluidos por los siguientes motivos: 13 casos por desarrollar la AHAI después de la realización de un trasplante de progenitores hematopoyéticos (TPH); y dos casos por imposibilidad de acceso a la historia clínica. El tamaño muestral final fue de 25 pacientes. Más de la mitad (72%) eran varones y el 28% mujeres. La mediana de edad al diagnóstico fue de dos años (rango de 0,4 a 9).
El 72% de los casos presentó un cuadro febril en los tres a 10 días previos a la aparición de la AHAI, y sólo el 44% asociaron clínica infecciosa. Otros síntomas frecuentes fueron los vómitos y/o dolor abdominal (40%). Tres pacientes (12%) debutaron con alteración del nivel de conciencia. La alteración en la coloración de piel y mucosas en forma de palidez o de ictericia fue el signo clínico predominante (68% y 64%, respetivamente), seguido de hepatoesplenomegalia (48%) y orina colúrica (48%). En un 32% se constató taquicardia y un 16% soplo cardíaco (tabla 1). De los 25 pacientes, 14 (56%) requirieron ingreso en unidad de cuidados intensivos para el manejo inicial por riesgo de inestabilidad hemodinámica.
Características de los 25 pacientes pediátricos con AHAI
| Variables | n (%) | |
|---|---|---|
| Sexo | • Varón | 18 (72) |
| • Mujer | 7 (28) | |
| Edad al diagnóstico | • < 1 año | 5 (20) |
| • 1 a 5 años | 16 (64) | |
| • 6 a 10 años | 4 (16) | |
| Signos/síntomas clínicos | • Fiebre | 18 (72) |
| • Infección respiratoria vías altas | 11 (44) | |
| • Dolor abdominal/Vómitos | 10 (40) | |
| • Alteración nivel conciencia | 3 (12) | |
| • Palidez | 17 (68) | |
| • Ictericia | 16 (64) | |
| • Hepatoesplenomegalia | 12 (48) | |
| • Hemoglobinuria | 12 (48) | |
| • Taquicardia | 8 (32) | |
| • Soplo cardiaco | 4 (16) | |
| Estudio inmunológico | • IgG positivo | 7 (28) |
| • IgG + C3d positivo | 10 (40) | |
| • C3d positivo | 6 (24) | |
| • IgA positivo | 1 (4) | |
| • Negativo | 1 (4) |
La mediana de Hb al diagnóstico fue de 5,4 g/dL (rango de 2,3 a 10,7). La cifra de reticulocitos estaba elevada en todos los casos salvo un paciente que presentó reticulocitopenia. Se detectó elevación de bilirrubina indirecta (92%), haptoglobina indetectable (88%) y LDH elevada (84%). Cinco pacientes desarrollaron otras citopenias durante su estudio. Dos de estos cinco pacientes se manifestaron inicialmente como AHAI y desarrollaron algún episodio de trombocitopenia inmune (PTI) a lo largo de su evolución; otros dos pacientes asociaban AHAI y PTI de forma concomitante en el momento del diagnóstico, y el caso restante debutó con una PTI y cinco años después presentó AHAI.
La mayoría de los casos (76%) fueron AHAI producidas por anticuerpos calientes (IgG o IgA) y el 24% restante por anticuerpos fríos. Dos pacientes tenían un TCD poliespecífico negativo. En el primer caso, se identificó positividad fuerte para IgA como anticuerpo responsable mediante la realización de un test monoespecífico con anticuerpos irregulares (Panocell Immucor®) en un centro especializado. En el segundo caso, se ampliaron estudios inmunohematológicos más complejos, se descartaron causas de anemia hemolítica hereditaria (electroforesis de hemoglobinas, fragilidad osmótica, autohemólisis, test EMA, cuantificación enzimática de glucosa-6-fosfato deshidrogenasa normales) y finalmente se objetivó respuesta al tratamiento empírico esteroideo empleado (tabla 1).
Se realizaron estudios inmunológicos y de autoinmunidad al diagnóstico a la mayoría de los pacientes (80%). Dichos estudios consistieron en la cuantificación de inmunoglobulinas, determinación de anticuerpos antinucleares y del complemento. Además, en aquellos pacientes que asociaron recaídas, citopenias y/o refractariedad al tratamiento esteroideo (10/25), se realizaron de forma complementaria y previo a la utilización de una segunda línea de tratamiento, estudios de subpoblaciones linfocitarias, apoptosis, detección de otros autoanticuerpos (anti-tiroideos, anti-DNA, anti-ENA, ANCA, antifosfolípidos) y detección de respuesta vacunal mediante la cuantificación del título de anticuerpos frente a tétanos, difteria y neumococo. Como resultado de esos estudios, sólo una paciente (4%), que asociaba además enfermedad intestinal y déficit de IgA, fue diagnosticada durante su evolución de inmunodeficiencia variable común (IDVC). Cinco pacientes (20%), todos ellos con AHAI por anticuerpos IgG, presentaron trombocitopenia concomitante (Síndrome de Evans, SE).
En ocho pacientes (32%), todos ellos AHAI por anticuerpos calientes, se sospechó o se documentó una infección viral en el momento del diagnóstico. Los principales microorganismos aislados mediante serología fueron: virus de Epstein Barr (VEB) en dos casos, citomegalovirus (CMV) en tres pacientes, un caso de parvovirus B19, uno de virus varicela zóster (VVZ) y otro de Mycoplasma pneumoniae.
La primera línea de tratamiento empleada fueron los corticoides en 24 pacientes (96%). El único caso que no recibió corticoides fue tratado con antibiótico intravenoso para la infección concomitante y una transfusión de hemoderivados por tratarse de AHAI por anticuerpos fríos. Se alcanzó RC en 18 pacientes (72%) y RP en seis pacientes (24%). El paciente restante presentó respuesta corticoidea inicial en el momento agudo, pero se desconoce la evolución completa por pérdida de seguimiento al ser trasladado a otro centro tras su estabilización inicial. Aquellos con RP recibieron otras líneas de tratamiento (rituximab, micofenolato de mofetilo [MFM], inmunoglobulinas, ciclosporina); 23 pacientes (92%) requirieron transfusión de hemoderivados durante el curso de la anemia.
Con respecto a los pacientes con AHAI producida por anticuerpos calientes (fig. 1), todos (19/19) recibieron tratamiento con corticoides inicialmente (2 mg/kg/día en tres dosis vía oral). De ellos, 11/19 (58%) alcanzaron RC tras una mediana de tratamiento corticoideo de tres meses (rango de 0 a cinco meses). El 42% restante (8/19), presentó RP y precisó otros tratamientos para alcanzar la RC. Dos de esos ocho pacientes recibieron tratamiento con rituximab intravenoso (375 mg/m2/semanal). Uno de ellos fue el paciente que presentó TCD negativo que respondió y alcanzó RC tras completar el ciclo de cuatro dosis de rituximab. El otro presentó una reacción alérgica grave con la tercera dosis y se realizó cambio a ciclosporina (90 mg/12 h vía oral durante cuatro meses). Coincidiendo con una nueva infección, precisó asociar al tratamiento con ciclosporina (50 mg/12 h), MFM (400 mg/8 h vía oral) durante cinco meses alcanzando finalmente RC a los 12 meses del diagnóstico. Cinco de los ocho pacientes con RP asociaron PTI (SE) y precisaron tratamiento con inmunoglobulinas (1 g/kg intravenoso) como coadyuvante al esteroide. Tres de esos cinco pacientes presentaron una evolución tórpida requiriendo múltiples líneas de tratamiento: MFM (500 mg/12 h), mercaptopurina (50 mg/m2/día), ciclosporina (50-150 mg/12 h), timoglobulina (3,75 mg/kg/día durante cinco días), vincristina (1-2 mg/día intravenosa), esplenectomía, e incluso TPH en dos de ellos. La mediana de seguimiento fue de 3,5 años (rango de 1 a 17). Un 26% (5/19) presentaron alguna recaída durante su evolución. Se detectaron complicaciones en seis pacientes (32%), principalmente infecciones de repetición, además de dos casos de litiasis biliar y un paciente con importante retraso del crecimiento como efecto secundario del tratamiento esteroideo prolongado. Se registró un único caso de exitus posterior a TPH.
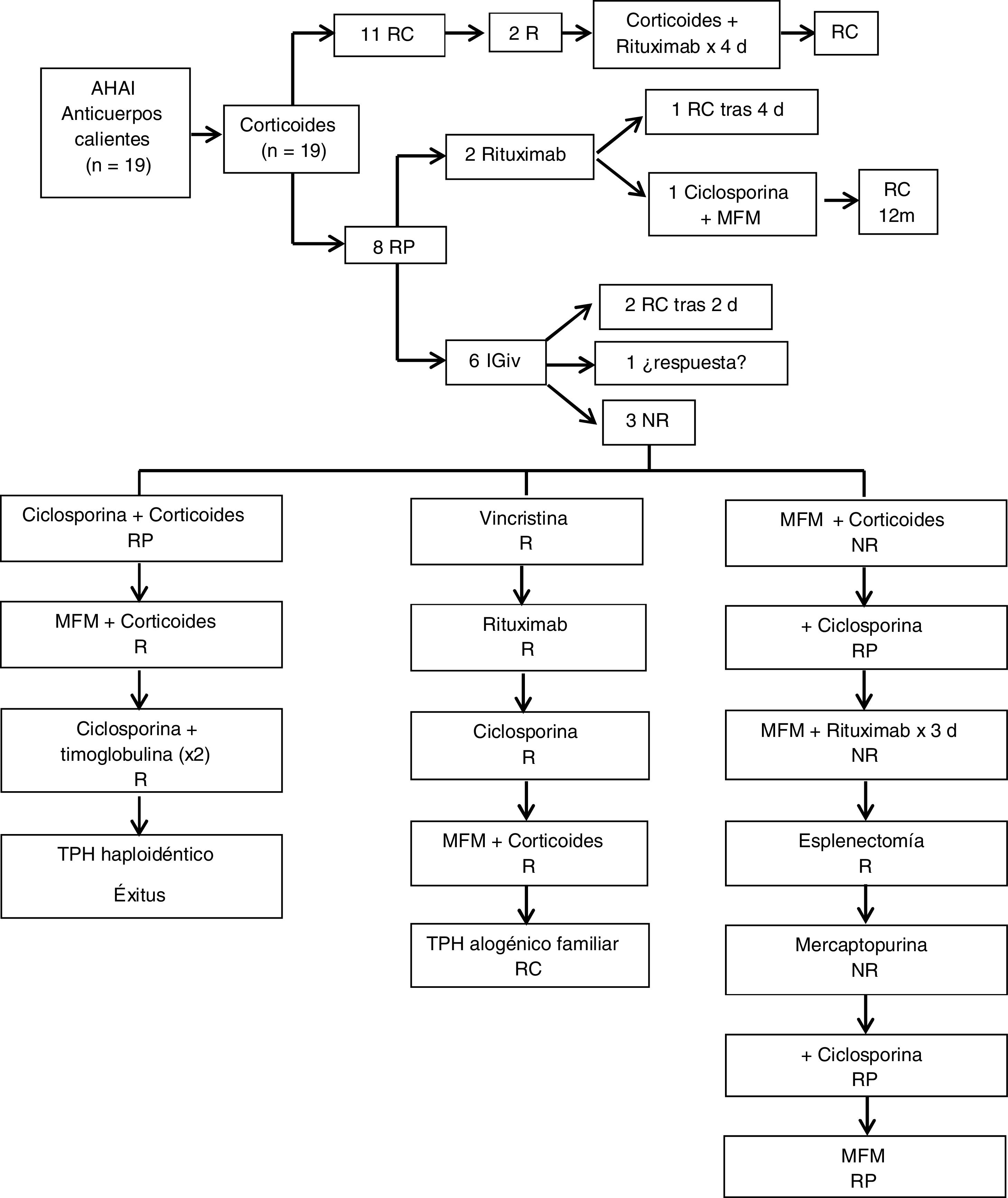
Tratamiento y respuesta en pacientes con anemia hemolítica autoinmune por anticuerpos calientes. RC: remisión completa, RP: respuesta parcial, NR: no respuesta, R: recaída, d: dosis, m: meses, IGiv: inmunoglobulina intravenosa, MFM: micofenolato de mofetilo, TPH: trasplante de progenitores hematopoyéticos.
En cuanto a los pacientes con AHAI producida por anticuerpos fríos (tipo IgM), 5/6 (83%) recibieron tratamiento con corticoides en dosis única. En el caso restante la anemia se resolvió tras el tratamiento de la infección concomitante y una transfusión de hemoderivados, como se ha indicado previamente. Todos alcanzaron RC. La mediana de seguimiento fue de tres meses (rango de 0 a siete meses). No se detectaron complicaciones ni recaídas durante el seguimiento (fig. 2).
Discusión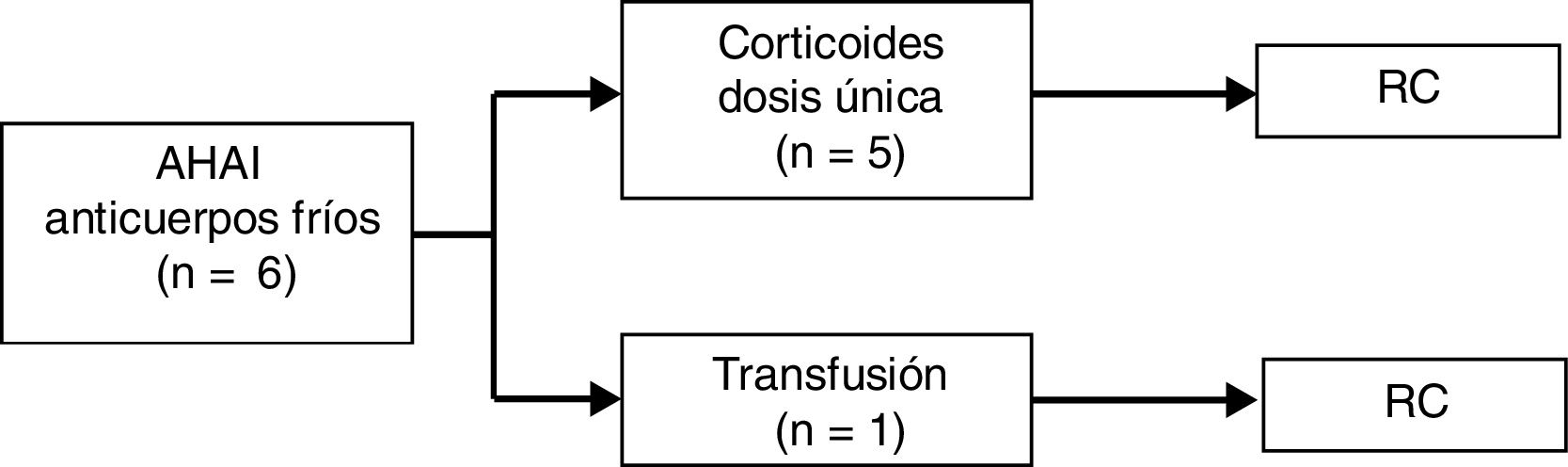
La AHAI es una entidad rara en pediatría, tal y como se confirma por la escasez de casos en nuestra serie retrospectiva que abarca más de 20 años. La mayoría de nuestros casos de AHAI fueron autolimitados y respondieron al tratamiento con corticoides (72%) por lo que, a pesar de su inicial gravedad, puede catalogarse como una enfermedad de curso benigno en un alto porcentaje de pacientes.
Tanto en niños como en adultos, la causa más frecuente de AHAI es aquella producida por anticuerpos calientes (70-80%)8,9, dato que se reproduce en nuestro estudio (76%). Tras el diagnóstico de anemia hemolítica, es importante descartar causas secundarias como responsables de la misma (trastornos linfoproliferativos, enfermedades autoinmunes, inmunodeficiencias primarias, fármacos o agentes infecciosos), puesto que en la mayoría de ellas la etiología es secundaria a un proceso subyacente10,11. En la actualidad, la mayoría de los algoritmos diagnósticos2,5 proponen realizar estudios de inmunidad y autoinmunidad a todos los pacientes en el momento del diagnóstico (anticuerpos antifosfolipídicos y antinucleares, cuantificación de inmunoglobulinas, subpoblaciones linfocitarias y complemento) y repetirlos anualmente aunque el resultado fuese normal, ya que la causa subyacente puede ponerse de manifiesto después del diagnóstico de AHAI. En nuestra opinión estos estudios deben incluirse en la evaluación inicial de todos los casos atípicos, en todos los pacientes refractarios y en el momento de la recaída. Además, según la historia clínica, los hallazgos en la exploración física y en las pruebas complementarias puede ser necesaria la realización de otros estudios como aspirado/biopsia de médula ósea, pruebas de imagen o citometría de flujo, en función de la sospecha diagnóstica.
Un alto porcentaje de los pacientes incluidos en el estudio presentaba historia de clínica infecciosa previa al debut de la AHAI, pero tan sólo se detectó aislamiento microbiológico concomitante en menos de la mitad de los casos. Los gérmenes aislados fueron fundamentalmente virus (CMV, VEB, parvovirus B19, VVZ) y Mycoplasma pneumoniae, principales responsables recogidos en la literatura12,13. La mayoría de estas infecciones están asociadas con crioaglutininas por autoanticuerpos IgM14. A pesar de que una causa frecuente de AHAI en población pediátrica son las infecciones virales, es difícil establecer si la propia infección es el desencadenante o el causante principal de dicha anemia.
Cinco pacientes de nuestra cohorte fueron diagnosticados de SE. A diferencia de los pacientes con AHAI aislada, los pacientes con SE suelen presentar un curso crónico, recurrente y refractario al tratamiento15,16. En nuestra serie ninguno de ellos alcanzó RC con el tratamiento esteroideo aislado, y precisan asociar otros tratamientos, confirmando este peor pronóstico. Un porcentaje significativo de casos con SE tienen un síndrome linfoproliferativo autoinmune (ALPS) subyacente17,18, por lo que es importante descartarlo en estos pacientes al igual que otras entidades autoinmunes e inmunodeficiencias, como se ha detallado anteriormente. En nuestra cohorte no hemos podido confirmar este diagnóstico en ningún caso. También se asocia a otras alteraciones reumatológicas e inmunológicas, como la IDVC19. En nuestro estudio, uno de los pacientes con SE fue diagnosticado a lo largo de su evolución de IDVC.
La presencia de positividad en el TCD confirma el diagnóstico de AHAI cuando se realiza el estudio de una anemia hemolítica adquirida. Sin embargo, hasta un 3-11% según las series, tienen TCD negativo20–22. La presencia de una IgG de baja afinidad, la sensibilización de IgG por debajo del umbral de detección del reactivo comercial o la existencia de anticuerpos tipo IgA que no son detectados en los TCD de rutina, son algunos de los principales motivos de AHAI con TCD negativo23–25. Cuando la sospecha clínica de AHAI sigue siendo alta a pesar de un TCD negativo, se debe considerar la realización de otros estudios inmunohematológicos más complejos en centros especializados26, así como la realización del test Donath-Landsteiner para descartar la presencia de una hemolisina bitérmica. Dos pacientes de nuestra cohorte presentaron TCD negativo y se amplió el estudio en laboratorios especializados. En uno de ellos se demostró IgA como anticuerpo responsable. En el segundo todos los estudios fueron negativos, pero se consideró una AHAI por anticuerpos calientes por su comportamiento clínico-terapéutico con tendencia a la recidiva tras retirada de corticoide. Cuando no es posible realizar otras técnicas o éstas no son concluyentes pero los hallazgos clínico-analíticos son compatibles con AHAI, una vez descartadas causas hereditarias de la misma, se debe establecer el mismo manejo terapéutico que los casos con TCD positivo26.
Dada la ausencia de ensayos clínicos aleatorizados y randomizados, la elección del tratamiento de pacientes pediátricos con AHAI está basada en series de casos y opiniones de expertos27.
Existe un acuerdo general de que los corticoides representan la primera línea de tratamiento en AHAI por anticuerpos calientes. Un 70-85% de los casos alcanzan respuesta únicamente con este tratamiento, con una mediana de tiempo de inicio de respuesta de 15 días (rango de una a tres semanas)28. Es importante hacer un descenso lento y progresivo siendo necesario en muchos casos prolongarlo hasta seis meses. Aproximadamente un 20-30% requerirá una segunda línea de tratamiento que se iniciará en casos de ausencia de respuesta o refractariedad corticoidea28. Algunos estudios apoyan el empleo de rituximab como fármaco de elección de segunda línea con una mediana de tiempo de inicio de respuesta de 1,4 meses (rango de 0,5 a cinco meses)29,30. Otros autores prefieren la esplenectomía como segunda opción con tasas de respuesta que oscilan entre un 60 y 90%31. Sin embargo, no existen estudios que comparen la eficacia entre los tratamientos disponibles como segunda línea y la esplenectomía. Además, no se recomienda realizar una esplenectomía en pacientes menores de seis años. La utilización de otras terapias como MFM, azatioprina, ciclosporina, inmunoglobulinas, alemtuzumab, se reservan para casos refractarios. No se conocen con claridad los índices de respuesta de estos fármacos y debido a ello, no existe consenso acerca de la superioridad de emplear un fármaco frente a otro32. Por tanto, la elección de la terapia a utilizar debe realizarse de forma individualizada en cada caso, según la respuesta hematológica alcanzada y los potenciales efectos secundarios farmacológicos. En las AHAI por anticuerpos fríos se recomienda tratamiento sintomático y de la causa subyacente33. Se debe reservar la transfusión para los casos de anemia grave o alteración de signos vitales del paciente34. En nuestra muestra, la gran mayoría de los pacientes recibieron al menos una transfusión. Los índices de respuesta son los habitualmente descritos en otras series. Es importante señalar que la respuesta no comienza a aparecer hasta después de las dos primeras semanas en las AHAI por anticuerpos IgG, no siendo recomendable el cambio de actitud terapéutica antes de alcanzarse al menos este período de tiempo. Los pacientes diagnosticados de SE requirieron varias líneas de tratamiento y, por tanto, como se ha reportado también previamente, es esperable en ellos una evolución más compleja.
La principal limitación de nuestro estudio es el pequeño tamaño muestral, la dificultad para definir el momento de realización de algunos estudios y la pérdida de seguimiento de algunos pacientes. Sin embargo, los resultados obtenidos en esta serie son similares a los descritos en otras series pediátricas. Por este motivo podemos indicar que contribuye a consolidar el conocimiento del comportamiento de la AHAI en niños.
Como conclusión de este estudio debemos indicar que como ya se ha señalado, la AHAI en la infancia es una entidad muy poco frecuente, con una evolución favorable en la mayoría de los casos al ser secundaria a procesos infecciosos. Sin embargo, en ciertos casos puede evolucionar de manera tórpida. En estos, es necesario el despistaje de entidades subyacentes más graves, como inmunodeficiencias o enfermedades inflamatorias, y el empleo de fármacos de segunda línea. Por este motivo, sería aconsejable que al menos todos estos casos de evolución desfavorable fuesen remitidos a centros con experiencia en el manejo de estos pacientes.
FinanciaciónEste trabajo no ha recibido ningún tipo de financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
