


El retraso mental afecta al 3% de la población. En el 50% no es posible determinar la etiología. Las alteraciones cromosómicas submicroscópicas subteloméricas, no detectables con técnicas citogenéticas convencionales, pueden explicar algunos casos de retraso mental criptogénicos.
Pacientes y métodosCohorte de 200 pacientes, con edades comprendidas entre los 2,5 y los 15 años, y retraso psicomotor (< 6 años) o retraso mental (> 6 años) criptogénicos. Variables: grado de retraso, dismorfias (faciales, manuales, macrosomía/microsomía), crecimiento intrauterino retardado, epilepsia. Identificación de reordenamientos cromosómicos subteloméricos mediante MLPA (multiplex ligation dependent probe amplification), que detecta pérdidas o ganancias de material genético. Confirmación de los hallazgos patológicos mediante FISH (fluorescent in situ hybridization) y/o array de CGH (comparative genomic hybridization).
ResultadosSe detectaron anomalías subteloméricas en 9 pacientes, lo que representa el 4,5% de los casos. El estudio de progenitores demostró en un caso una traslocación en equilibrio. El resto eran alteraciones «de novo». Existía asociación significativa con la presencia de más de un rasgo fenotípico dismórfico o el antecedente de crecimiento intrauterino retardado, pero no con el grado de retraso ni con la presencia de epilepsia.
ConclusionesLas alteraciones cromósomicas submicroscópicas subteloméricas explican el 4,5% de los retrasos mentales de causa desconocida en nuestra serie. En nuestra población se asocian a la presencia de más de un rasgo fenotípico anormal o al antecedente de crecimiento intrauterino retardado.
Mental retardation affects 3% of the population, the origin of which cannot be established in 50% of cases. Subtelomeric rearrangements, not detected by routine cytogenetic studies, might explain some cases of unknown cause.
Patients and methodsA study was conducted on 200 subjects with unexplained mental retardations using multiplex ligation dependent probe amplification (MLPA). Abnormal findings were confirmed by fluorescent in situ hybridization (FISH) and/or comparative genomic hybridization technology (CGH-array).
ResultsA subtelomeric aberration was identified in 9 patients. Eight were «de novo»; one was inherited from a phenotypically normal parent. There was a statistically significant association with the presence of more than one dysmorphic feature or with intrauterine growth retardation, but not with the severity of retardation or epilepsy.
ConclusionsSubtelomeric rearrangements explained 4.5% of cases of mental retardation in our series. The presence of more than one dysmorphic feature or intrauterine uterine growth retardation increases the probability of this type of chromosomal aberration.
El retraso mental (RM) afecta al 3% de la población1, cifra que orienta acerca de la trascendencia clínica y social del problema. Las causas del RM son muy variadas y con los métodos de diagnóstico rutinarios disponibles actualmente es posible establecer la etiología en el 40-50% de los casos. Entre estos, dos terceras partes son debidos a causas adquiridas, como lesiones cerebrales hipóxicas, infecciosas o tóxicas pre, peri y posnatales, malformaciones del sistema nervioso central, errores innatos del metabolismo o privación ambiental grave. El tercio restante se debe a alteraciones cromosómicas. Estas representan, por lo tanto, una causa importante de RM, detectándose hasta en un 40% de los casos de RM grave o un 10% de los casos de RM moderado a leve. En el 50-60% de los casos de RM catalogados como «criptogénicos» se considera que causas genéticas no bien conocidas desempeñan un papel importante2. Esta alta tasa de casos en los que no es posible determinar la etiología limita tanto el diagnóstico etiológico como la eficiencia del consejo genético, e impide la detección de portadores y el diagnóstico prenatal en familias que tienen uno o más miembros afectados de RM.
En la última década se ha constatado que los reordenamientos cromosómicos submicroscópicos que afectan a los telómeros pueden ser una causa significativa de RM esporádico y familiar, con o sin malformaciones asociadas, explicando entre un 5% y un 30% de los RM sin filiar o criptogénicos2-10. Esto supone una mayor frecuencia en comparación con condiciones bien conocidas causantes de RM, como el síndrome de X frágil. Con estos antecedentes, se ha realizado un estudio orientado a identificar reordenamientos cromosómicos subteloméricos no detectables con las técnicas citogenéticas convencionales en pacientes de edad pediátrica con retraso psicomotor global (RPM) o RM. Otros objetivos del estudio han sido el establecimiento de correlaciones fenotipo-genotipo en individuos en los que se hayan detectado anomalías subteloméricas y analizar algunas variables clínicas como indicadores de alteraciones de este tipo.
Sujetos y métodosSujetos de estudioCohorte de 200 pacientes, de edades entre 2,5 y 15 años, diagnosticados de RPM (< 6 años) o RM (> 6 años) criptogénico. Los pacientes fueron reclutados entre enero de 2007 y diciembre de 2009 en las unidades de neurología pediátrica de los hospitales Virgen de la Salud (Toledo) y General de Guadalajara. La muestra se compuso de pacientes en seguimiento y de pacientes remitidos durante el periodo de reclutamiento desde los centros base de bienestar social, centros de atención primaria, unidades de salud mental o gabinetes de orientación pedagógica escolares. En todos los casos fueron normales los antecedentes y/o las exploraciones destinadas a descartar otras etiologías del RPM o RM. Ninguno de los casos presentaba criterios de exclusión: a) enfermedad neurológica por patología cerebral adquirida (encefalopatías hipóxico-isquémicas en sentido amplio, parálisis cerebral infantil, infección del SNC, traumatismo craneoencefálico grave, evidencia demostrable o retrospectiva de daño cerebral pre/perinatal/postnatal de origen infeccioso o tóxico); b) síndrome genético con afectación del SNC (cromosomopatías, X-frágil, síndromes por genes contiguos, síndromes de causa genética establecida, síndromes neurocutáneos, encefalopatías degenerativas); c)encefalopatías metabólicas (errores innatos del metabolismo intermediario, enfermedades lisosomales y peroxisomales, citopatías mitocondriales, esfingolipidosis y enfermedades por depósito de macromoléculas), y d)privación emocional, ambiental, afectiva y sensorial. Se obtuvo el consentimiento informado de los padres o tutores legales para la participación en el estudio. La investigación fue aprobada por los comités éticos de los centros hospitalarios participantes.
Variables clínicasEn todos los casos se determinaron las siguientes variables: a)grado de RPM o de RM. Mediante los tests WPPSI o la escala de Bayley de desarrollo infantil (< 6 años) o WISC-IV (> 6 años), que permiten valorar el cociente intelectual (CI) o el cociente de desarrollo (CD). El grado de RM se clasificó según los criterios DSM-IV (Diagnostic Statistic Manual de la Academia Americana de Psiquiatría) en leve (CI 50-70), moderado (CI 35-50) y grave (CI inferior a 35). Se utilizó un criterio similar para la clasificación del CD en los niños con RPM de edad inferior a 6 años; b) rasgos fenotípicos anormales. Se seleccionaron, basándose en datos de estudios publicados previamente4–6, los siguientes: microcefalia/macrocefalia y alteraciones de la forma craneal, macrosomía (peso y talla>P97), microsomía (peso y talla<P3), hipertelorismo/anomalías palpebrales, anomalías nasales y faciales (filtrum, boca, labios, etc.), anomalías en pabellones auriculares, anomalías en manos/dedos; c) presencia o ausencia de crecimiento intrauterino retardado (CIR), y d) presencia o ausencia de epilepsia.
Estudios genéticosSe utilizaron muestras de sangre periférica. Previamente, se realizó cariotipo de alta resolución, que fue normal en todos los casos. El rastreo de desequilibrios cromosómicos subteloméricos se llevó a cabo mediante la técnica de MLPA (multiplex ligation dependent probe amplification) empleando los kits comerciales SALSA P036 Y P070 (MRC-Holland, Amsterdam, Holanda), de acuerdo con las instrucciones del fabricante disponibles en http://www.mlpa.com.
Los productos resultantes de la reacción de MLPA se sometieron a electroforesis capilar en un analizador ABI 310 (Applied Biosystems) y se analizaron con el software GeneMapper. En aquellos casos en los que se reveló la presencia de un desequilibrio cromosómico subtelomérico con ambos juegos de sondas, se analizó una muestra de los progenitores siguiendo la misma metodología para descartar la existencia de un polimorfismo heredado. Para confirmar los desequilibrios cromosómicos no heredados detectados mediante MLPA, se empleó un segundo método: FISH, CGH Array, o ambos. El FISH (fluorescent in situ hybridization) es una técnica citogenética que permite demostrar la presencia o ausencia de una región cromosómica concreta. En nuestro estudio se hibridó una extensión de metafases del paciente con sondas subteloméricas comerciales (Poseidon, Kreatech Diagnostics, Amsterdam, Holanda) correspondientes a la región en desequilibrio y de acuerdo con las instrucciones del fabricante. El CGH Array (comparative genomic hybridization) es una técnica de genética molecular que permite demostrar ganancias o pérdidas de material genético de un paciente frente a un control en todo el genoma o parte de él. La muestra de ADN del paciente se hibridó con ADN comercial de referencia y de igual sexo (Promega Biotech) sobre una plataforma de array-CGH de 44.000 oligonucleótidos distribuidos a lo largo de todo el genoma (Human 44K Agilent Technologies, Santa Clara, EE. UU.) de acuerdo con las instrucciones del fabricante. De los dos métodos descritos, el FISH es el más adecuado por su coste y escasa complejidad técnica. No obstante, en algunos de nuestros casos se optó por el CGH Array con el fin no solo de confirmar sino de definir exactamente el tamaño del desequilibrio.
Bases de datos públicasPara establecer una posible correlación entre las alteraciones subteloméricas detectadas y los datos clínicos de otros pacientes con alteraciones similares se ha recurrido a las bases de datos ECARUCA y DECIPHER. Para investigar las posibles implicaciones clínicas de las alteraciones detectadas en relación con los genes que se ven afectados se ha empleado la base de datos Ensemble.
Análisis estadísticoCon los datos del estudio se realizó un análisis estadístico empleando para determinar si alguna de las variables clínicas analizadas (grado de RPM/RM-leve vs. moderado/grave-profundo, presencia de más de un rasgo fenotípico anormal vs. uno o ausencia de rasgos fenotípicos, presencia vs. ausencia de CIR, presencia vs. ausencia de epilepsia) era un indicador sensible de la presencia de reordenamientos crípticos subteloméricos. Se establecieron tablas de contingencia de 2 x 2 y análisis mediante la prueba de Fisher. Se consideró un nivel de significación estadística p<0,05.
ResultadosLos datos globales de la población de estudio en relación con el grado de RPM/RM, rasgos fenotípicos, CIR y epilepsia fueron los siguientes: RPM/RM leve 52%, moderado 32%, grave-profundo 16%; más de un rasgo fenotípico anormal 33%, uno o ningún rasgo fenotípico 67%; CIR 8%, ausencia de CIR 92%, y presencia de epilepsia 20%, ausencia de epilepsia 80%.
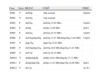
De los 200 pacientes del estudio, en 12 (6%) se detectó mediante MLPA alguna alteración en el número de copias de alguna de las regiones subteloméricas (tabla 1). En 7 pacientes se detectó una deleción como única alteración y en otros 4 pacientes se hallaron simultáneamente una deleción y una duplicación en distintos brazos cromosómicos. Por tanto, mediante MLPA se detectaron un total de 15 alteraciones en 11 pacientes distintos. En 2 pacientes (RM1 y RM2) la reducción en el número de copias correspondientes a los brazos 20p y 19q, respectivamente, observada por MLPA no se confirmó por FISH o array de CGH, revelando ambas técnicas la existencia de dos copias en cada una de las zonas subteloméricas mencionadas. En 10 pacientes (RM3-RM12), las técnicas de FISH y/o array de CGH confirmaron los hallazgos del MLPA. En la tabla 2 se muestran los datos del grado de RPM/RM, rasgos fenotípicos, CIR, epilepsia y otras características en los 10 casos que presentaron alteraciones submicroscópicas subteloméricas confirmadas por FISH y/o array de CGH. No obstante, el caso RM9 se ha excluido como positivo, ya que al introducir la región perdida en la base de datos DECIPHER y en la de variantes genómicas humanas encontramos que no parece contener ningún gen de relevancia que pueda relacionarse con la clínica y que aparece descrita en numerosos controles normales como una variante polimórfica. Por ello es posible que se trate de un hallazgo no relacionado con el retraso del paciente, aunque tampoco puede descartarse. En resumen, se confirmaron anomalías por FISH y/o array de CGH en 9 de los 12 casos con hallazgos anormales por MLPA, lo que representa que se encontraron verdaderos reordenamientos subteloméricos en el 4,5% de la población estudiada. En el estudio de los progenitores de los casos afectados se ha encontrado que solo en un caso (RM8) uno de los progenitores presentaba una traslocación en equilibrio. En el resto de los casos se trataba de alteraciones «de novo».
Resumen de las alteraciones detectadas por MLPA y su confirmación o no por FISH y/o CGH
| Caso | Sexo | MLPAa | CGHb | FISHc |
| RM1 | V | del20p | 20p normal | 20pX2 |
| RM2 | V | del19q | 19q normal | |
| RM3 | F | del22q | del22q (7,92 Mb) | 22qX1 |
| RM4 | F | del4p | del4p (1,89 Mb) | 4pX1 |
| RM5 | F | del22q | del22q (0,74 Mb) | 22qX1 |
| RM6 | F | del22q/dup20q | del22q (1,97 Mb)/dup20q (3,88 Mb) | 22qX1 |
| RM7 | V | dup15q | dup15q (5,83 Mb) | |
| RM8 | F | del10q/dup22q | del10q (8,6 Mb)/dup22q (3,43 Mb) | |
| RM9 | V | del17p | del17p (0,14 Mb) | |
| RM10 | V | del8p/dup4p | del8p (8,61 Mb)/dup4p (7,11 Mb) | |
| RM11 | F | del4p/dup19p | del4p (1,43 Mb)/dup19p (4,06 Mb) | 4pX1 |
| RM12 | V | del 1p | 1p X1 |
Datos clínicos principales de los casos que presentan alteraciones submicroscópicas subteloméricas confirmadas por FISH y/o CGH
| Caso | Sexo | Grado de RM/RPM | Rasgos fenotípicos principales | CIR | E | Otros |
| RM3 | F | Grave | Raíz nasal ancha, hendiduras palpebrales estrechas, macrosomía somática | No | No | Lenguaje ausente, hipotonía |
| RM4 | F | Moderado | Microcefalia, nariz larga, filtrum corto, comisuras bucales hacia abajo, frente prominente | Sí | Sí | Lenguaje presente |
| RM5 | F | Leve | Macrosomía somática | No | No | Lenguaje presente, hipotonía |
| RM6 | F | Moderado | Hipertelorismo, anomalías pabellones auriculares, filtrum largo, retraso crecimiento posnatal | No | No | Lenguaje ausente, rasgos autistas, hipotonía |
| RM7 | V | Grave | Estrechamiento craneal bitemporal, macrostomía, macroftalmia, retraso crecimiento posnatal | No | No | Lenguaje ausente, ausencia contacto visual |
| RM8 | F | Moderado | Epicantus, boca en «V» invertida, labios finos clinodactilia | Sí | No | |
| RM9 | V | Grave | Microcefalia | No | Sí | Lenguaje ausente, rasgos autistas |
| RM10 | V | Leve | Labios gruesos, filtrum largo macrosomía somática | No | No | Atrofia óptica |
| RM11 | F | Moderado | Anomalías pabellones auriculares, hipoplasia facial media, microsomía somática | Sí | Sí | Lenguaje ausente, hipomimia facial, hipotonía |
| RM12 | V | Moderado | Braquicefalia, puente nasal plano, pabellones auriculares desplegados | No | Sí | Espasticidad en miembros inferiores |
CIR: crecimiento intrauterino retardado; E: epilepsia; F: femenino; RM/RPM: retraso mental/retraso psicomotor; V: masculino.
El análisis estadístico demostró una asociación significativa en caso de presentar más de un rasgo fenotípico anormal y en caso de existencia de CIR. No se encontró asociación con el grado de RM/RPM ni con la presencia de epilepsia (tabla 3).
Asociación entre la presencia de anomalías submicroscópicas subteloméricas y las principales variables clínicas del estudio
| Variable clínica | Significación estadísticaa |
| > 1 rasgo dismórfico vs. ≤ 1 rasgo dismórfico | p<0,01 |
| CIR vs. ausencia CIR | p=0,02 |
| RM/RPM moderado-grave vs. leve | p=0,22 |
| Epilepsia vs. ausencia de epilepsia | p=0,16 |
El RM (o RPM en niños menores de 6 años) afecta aproximadamente a un 3% de la población. A un porcentaje considerable de estos pacientes y sus familias no es posible ofrecerles un diagnóstico etiológico o una explicación del problema. Los objetivos de nuestro estudio han sido determinar en qué grado las alteraciones submicroscópicas subteloméricas eran causa de RM/RPM y qué relación podría establecerse entre las alteraciones que se hallaran y las manifestaciones fenotípicas.
Con respecto a su prevalencia, hemos encontrado un porcentaje similar al de otras series descritas en la literatura, con cifras alrededor del 5%2-6,9,11-20. Queremos hacer notar que nuestra población de estudio no está especialmente seleccionada, siendo el criterio de inclusión la presencia de RM/RPM en cualquier grado, de causa desconocida, independientemente de la presencia de dismorfias u otras alteraciones fenotípicas. En series en las que se encuentra un porcentaje mayor de este tipo de alteraciones se seleccionaron pacientes con RM asociado a un número variable de rasgos dismórficos4,10,21-25. En cuanto al tipo de alteraciones detectadas, llama la atención el número de casos con deleción en 22q terminal, 3/11 (25%) y desequilibrios en 4p terminal (3/11) (25%). Al comparar estas cifras con las descritas en la base de datos ECARUCA, hemos constatado que se encuentran entre el tipo de alteraciones más frecuentes junto con la deleción en 1p terminal, de la que también hemos detectado un caso. Así pues, nuestra población de estudio no parece diferir de lo descrito en la literatura, ni en la proporción ni en el tipo de alteraciones encontradas.
Las correlaciones genotipo-fenotipo en estas alteraciones genéticas son interesantes pero complejas. En nuestra serie hay 3 pacientes, RM3, RM5 y RM6, que presentan deleción terminal de 22q. La pérdida de 22q13.3 es una de las deleciones con mayor incidencia en las bases de datos públicas que hemos manejado y se la considera responsable del síndrome de Phelan-Mc Dermid26. Los tres muestran características propias del síndrome, como retraso grave del desarrollo del lenguaje, hipotonía y macrosomía en dos de ellos (RM3 y RM5). Sin embargo, la paciente RM6 no solo no presenta macrosomía, sino que ha desarrollado un retraso de talla y peso posnatal. Esta paciente, además de la deleción 22q13, presenta una duplicación en el brazo p del cromosoma 20 y al revisar en ECARUCA casos con dup 20 p encontramos en varios de ellos retraso del crecimiento o talla baja, así como rasgos faciales comunes con la paciente RM6. El principal gen de relevancia clínica que se demuestra que se ha perdido en estos 3 pacientes es SHANK3. SHANK3 codifica para una proteína estructural involucrada en la maduración y la estabilización de las sinapsis entre neuronas, y la haploinsuficiencia de este gen, por deleción o mutación, es considerada responsable de los síntomas neurológicos del síndrome de Phelan-Mc Dermid27-29. De modo que en estos 3 casos podemos atribuir las manifestaciones clínicas de los pacientes a las alteraciones descubiertas en su genotipo y proporcionar un diagnóstico. En la revisión de la literatura no puede establecerse con claridad si existe una relación entre el tamaño de la deleción 22q13.3 y la clínica de los pacientes. Los datos que podemos aportar a este respecto son los siguientes: la paciente RM3 presenta una gran deleción de 7,92 Mb y sufre un RM profundo, ausencia total del lenguaje y grave hipotonía, mientras que la deleción presente en RM5 es unas 10 veces menor (0,74 Mb) y presenta un RM mental leve y ligera hipotonía. Por lo tanto, en nuestros casos el tamaño de la deleción sí parece estar relacionado con la gravedad del fenotipo.
Dos pacientes, RM4 y RM11, presentan deleción de 4p terminal de 1,8 Mb y 1,4 Mb, respectivamente. Es especialmente interesante el caso RM4, que presenta una deleción en 4p16.3, a cuya pérdida se atribuye el síndrome de Wolf-Hirschhorn30. Su fenotipo coincide solo en parte con los propios de este síndrome, pero a la vista del material perdido se observa que la deleción comienza justo en el límite del lugar donde se halla uno de los genes candidatos, WSCH2, sin que se pueda asegurar que este se halla perdido. Se demuestra, en cambio, la pérdida del gen LETM1, al que se considera causante de las convulsiones que se presentan en el WHS31 y que también se manifiestan en esta paciente, de modo que esta paciente se ha diagnosticado como WHS no típico. La segunda paciente, RM11, no tiene perdidos los genes WSCH1 ni WSCH2, y no muestra las características fenotípicas propias del síndrome de Wolf-Hirschhorn, pero ha perdido, sin embargo, LETM1 y presenta crisis epilépticas.
Otro caso en el que se ha podido proporcionar un diagnóstico es el paciente RM12, que presenta una deleción terminal de 1p, confirmada por FISH. La deleción subtelomérica hallada en el paciente está bien documentada en las bases de datos y en la literatura32-37. La monosomía de 1p36 es la causante del síndrome de microdeleción terminal más frecuente. Las manifestaciones fenotípicas más relevantes son microcefalia, retraso del crecimiento y puente nasal plano. Nuestro caso, sin embargo, presenta una leve espasticidad en los miembros inferiores, rasgo no descrito previamente.
El caso RM7 presenta una duplicación 15q con un tamaño de 5,83 Mb y que abarca la región 15q11.2-15q13.1. En esta región, conocida por su inestabilidad, se encuentran los genes cuya deleción se asocia a los síndromes de Angelman y Prader-Willi, así como varios genes que codifican diversas subunidades del receptor GABA-A. Los fenotipos descritos en casos con dup15q subtelomérica, tanto en la base de datos ECARUCA como algunos publicados38-40, incluyen rasgos dismórficos craneofaciales, hipotonía y retraso psicomotor grave, al igual que nuestro caso, así como epilepsia y rasgos autistas en aproximadamente la mitad de los casos. Nuestro caso no ha presentado crisis epilépticas, pero la ausencia de expresividad facial, contacto visual y lenguaje son predictores de una conducta autista en el futuro.
Los casos RM8 y RM10 presentan del10q/dup22q y del8p/dup4p, respectivamente. La presencia de una combinación deleción/duplicación sugiere la presencia de un cromosoma derivado de una traslocación en equilibrio en uno de los progenitores, de modo que el primer paso es determinar el origen de novo o heredado del cromosoma alterado, hecho de gran importancia a la hora de poder ofrecer un consejo genético. Así, en el caso RM8, el origen es heredado de uno de los progenitores, mientras que en los casos RM10 y los casos RM6 y RM11 (comentados previamente) el origen es «de novo». En estos casos con alteraciones complejas no podemos establecer una correlación genotipo-fenotipo, ya que tratamos con más de una alteración cromosómica y ninguna de ellas por separado es causante de un síndrome descrito y bien definido, como es el caso de del22q, del4p o del1p. Al comparar en las bases de datos o en la literatura los fenotipos de los pacientes con otros que compartan alguna de las alteraciones cromosómicas, encontramos que estas, a su vez, se encuentran generalmente formando parte de otros reordenamientos complejos, de modo que no es posible atribuir un rasgo fenotípico concreto a una de las alteraciones cromosómicas. Es de destacar que el caso RM10 presenta una atrofia óptica congénita, hallazgo no descrito previamente en casos con del8p o dup4p.
Al analizar el valor de las variables clínicas (grado de RM/RPM, dismorfias, CIR, epilepsia) como indicadores de posible presencia de anomalías cromosómicas submicroscópicas subteloméricas, hemos encontrado una relación estadísticamente significativa con la existencia de dismorfias y el antecedente de CIR. Sin embargo, no hemos encontrado en nuestra población una asociación significativa con la presencia de epilepsia ni con el grado de RM/RPM.
Como conclusiones de los hallazgos encontrados en nuestra serie de pacientes podemos considerar:
- -
Las alteraciones cromósomicas submicroscópicas subteloméricas se encuentran detrás de algunos RM/RPM de causa desconocida. Su detección permite establecer un diagnóstico y ofrecer consejo genético a las familias mediante estudios en los padres para determinar si se trata de alteraciones «de novo» (esporádicas) o bien heredadas al ser uno de los progenitores portador de traslocación balanceada.
- -
La prevalencia de las anomalías cromósomicas submicroscópicas subteloméricas con significado claramente patogénico en nuestra población de estudio ha sido del 4,5%, porcentaje similar al encontrado en la literatura.
- -
Las anomalías cromósomicas submicroscópicas subteloméricas en nuestra población de estudio se asocian de forma estadísticamente significativa a los casos que presentan más de un rasgo fenotípico anormal (dismorfias craneofaciales, manuales, macrosomía/microsomía) o CIR.
- -
El análisis de las anomalías genéticas subteloméricas y los fenotipos asociados permitirá ampliar nuestro conocimiento sobre las correlaciones genotipo-fenotipo y el papel de algunos genes en pacientes con retraso psicomotor y mental. Para este fin es tan importante la consulta de las bases de datos clínicas y genómicas como el «vertido» de los resultados de los estudios para enriquecer las bases de datos actualmente existentes.
Los autores declaran no tener ningún conflicto de intereses.
FinanciaciónEl estudio ha sido financiado con la ayuda económica de una beca de investigación del FISCAM (Fondo de Investigación Sanitaria de Castilla-La Mancha), n.° 06043-00.
