



Las aplasias medulares congénitas constituyen un grupo heterogéneo de enfermedades que se caracterizan por insuficiencia medular, asociadas frecuentemente a una o más anomalías somáticas y con riesgo elevado de neoplasias. Son enfermedades raras, generalmente diagnosticadas en la edad pediátrica, y con una mortalidad prematura importante.
Los autores presentan 11 casos de aplasia medular congénita, 8 de anemia de Fanconi y 3 de disqueratosis congénita. Estos casos fueron diagnosticados en los últimos 14 años en el Hospital de Dona Estefânia.
The inherited aplastic anaemias are a heterogeneous group of disorders characterized by bone marrow failure, frequent association with one or more somatic anomalies and increased risk of cancer. They are rare disorders, usually diagnosed at paediatric age, and have significant premature mortality.
The authors report 11 cases of inherited aplastic anaemias, 8 of Fanconi′s anaemia and 3 of Dyskeratosis congenita. These cases were diagnosed in the last 14 years in the Dona Estefânia Hospital.
Las aplasias medulares son un grupo heterogéneo de enfermedades que se caracterizan por incapacidad de la médula ósea de producir un número adecuado de células de las 3 líneas hematopoyéticas. En un 15 a 20% de los casos tienen etiología congénita1.
Las aplasias medulares congénitas se asocian frecuentemente a una o más anomalías somáticas y tienen un riesgo aumentado de neoplasias. En la mayoría de los casos el diagnóstico se realiza en la edad pediátrica, sin embargo algunos son diagnosticados en la edad adulta y, actualmente muchas niños sobreviven en la edad adulta1,2. Los 2 síndromes de aplasia medular congénita más frecuentes son la anemia de Fanconi (AF) y la disqueratosis congénita (DC).
Casos clínicosEn los últimos 14 años, fueron diagnosticados, en el Hospital de Dona Estefânia, 11 casos de aplasia medular congénita, de los cuales, 8 de AF e 3 de DC.
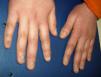
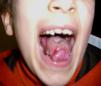
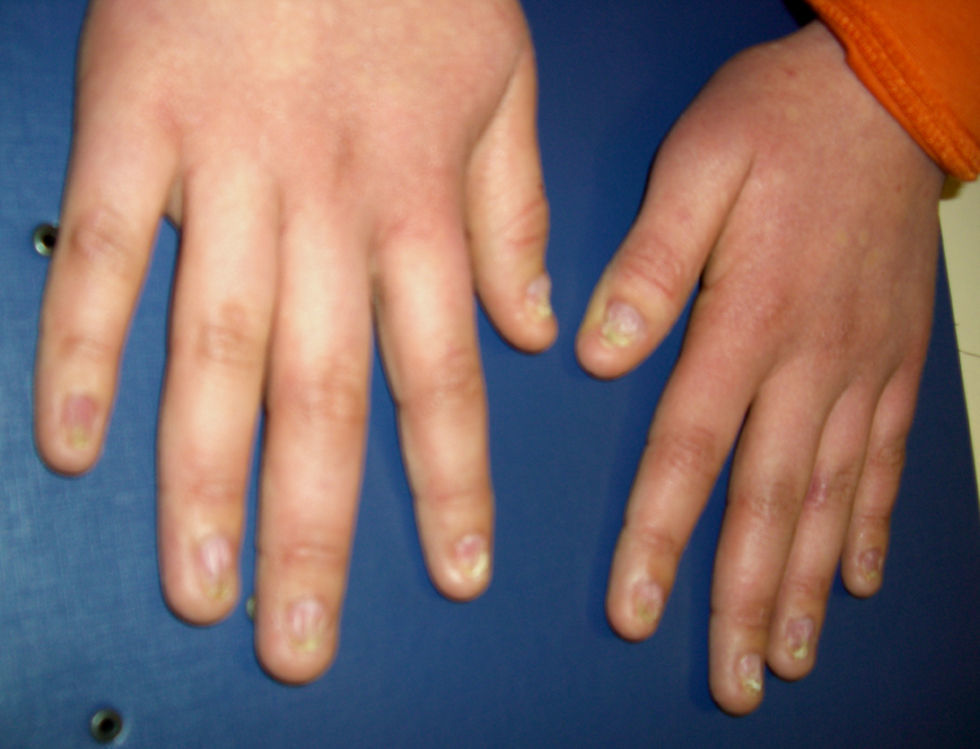
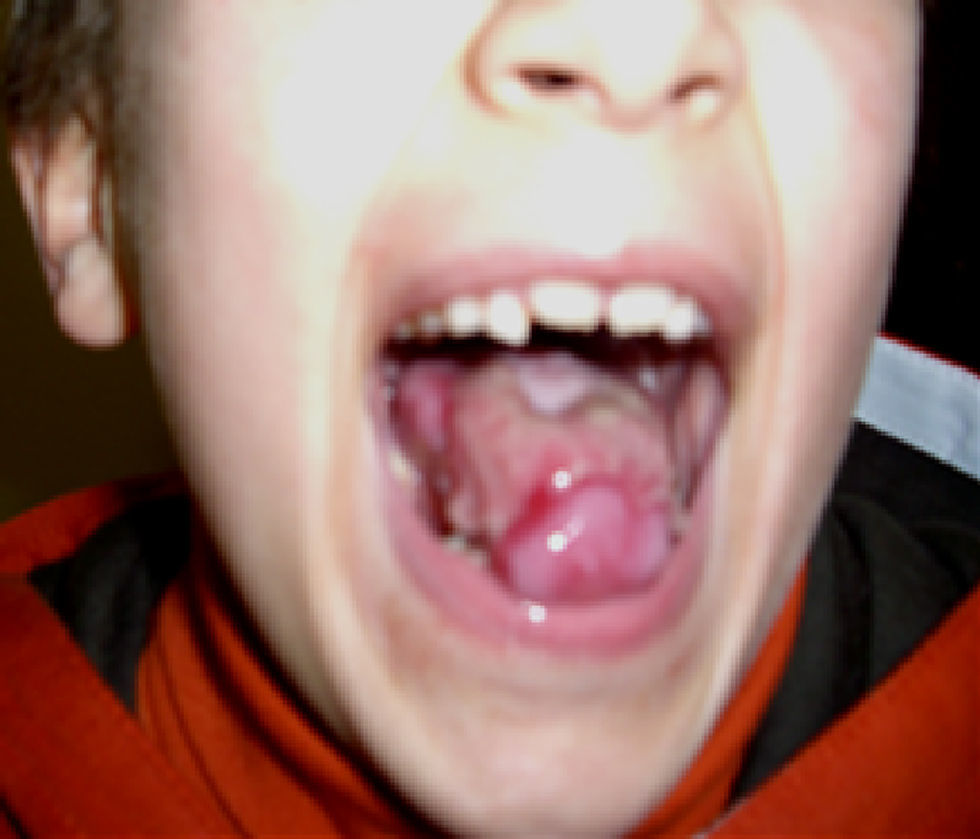
La media de edad en el momento del diagnóstico fue de 5 años para la AF y de 6 años para la DC. En los pacientes con AF, la relación entre el sexo masculino y femenino fue 1:1,6. Todos los pacientes con DC fueron del sexo masculino. Todos los pacientes con AF, exceptuando uno, tenían malformaciones congénitas (tabla 1). Los 3 pacientes con DC tenían la triade de manifestaciones mucocutáneas característica: hiperpigmentación reticular de la piel, distrofia de las uñas (fig. 1) y leucoplasia oral (fig. 2); un paciente tenía una estenosis del conducto lagrimal.
En lo que se refiere a las alteraciones hematológicas, solo en 2 pacientes con AF no fue posible identificar macrocitosis. Una paciente había realizado varias transfusiones sanguíneas antes de ser transferida de su país de origen y, otra paciente no realizó análisis y fue negado, por sus padres, seguimiento médico. En esta última paciente el diagnóstico de AF fue establecido a través de las manifestaciones somáticas y de su historia familiar. En el momento del diagnóstico todos los 10 pacientes estudiados tenían insuficiencia medular (7 pancitopenia, uno bicitopenia y 2 una citopenia aislada) y todos efectuaron estudios de fragilidad cromosómica en el ADN de linfocitos periféricos con diepoxibutano y mitomicina C (compatibles con AF en 7 pacientes). Efectuaron estudios genéticos 7 pacientes: en los cuatro pacientes con AF fue identificado el grupo de complementación FANC-A en 3, y FANC-G en uno; en los 3 pacientes con DC, fue identificada la misma mutación en el gen DKC1 (hemizigotia) en 2 hermanos y no fue identificada mutación en un paciente.
Ocho pacientes realizaron tratamiento con andrógenos. Los andrógenos utilizados fueron la oximetolona y, posteriormente, a partir de 2005, la nandrolona. De los 7 pacientes con AF, 5 tuvieron una buena respuesta una de ellas fue tardía, uno tuvo una respuesta parcial y, otro no obtuvo respuesta. El paciente con DC tuvo una buena respuesta medular. En relación a los efectos secundarios de los andrógenos se comenta: 5 pacientes tuvieron aumento de transaminasas hepáticas (transitoria n=4, persistente n=1), 6 pacientes tuvieron acné, 3 niñas presentaron signos de virilización, un niño tuvo aceleración de los caracteres sexuales secundarios. En el paciente con DC, fue necesario suspender el tratamiento por toxicidad hepática.
Cuatro pacientes necesitaron transfusiones regulares con concentrado de hematíes y plaquetas. Cuatro pacientes fueron tratados con factores estimuladores de colonias granulocíticas (FEC-G), en situación de infección.
Hasta el momento ningún paciente realizó transplante de células progenitoras hematopoyéticas. En dos pacientes con AF se encontró donante HLA compatible, pero en uno fue diagnosticado un tumor sólido antes del transplante y, otro falleció en la fase de condicionamiento para el transplante.
Respecto a la evolución clínica se comenta: 2 pacientes con DC no precisan ningún tratamiento, 4 pacientes con AF se encuentran en tratamiento con andrógenos con buena respuesta, una paciente con AF esta dependiente de régimen transfusional regular, un paciente con AF desarrollo un tumor sólido (base de la lengua); 4 pacientes fallecieron por complicaciones de la insuficiencia medular, 3 con AF e uno con DC.
DiscusiónLa AF fue descrita por primera vez en 1927 por un pediatra suizo, Guido Fanconi; desde entonces existen más de 1.850 casos descritos en la literatura2. Como se verificó en los casos clínicos presentados, existe una gran variedad clínica entre los pacientes que pueden inclusive no tener signos físicos de mayor1,10,11. Respecto a las manifestaciones hematológicas, todos los pacientes tenían macrocitosis y la pancitopenia fue la forma de presentación más común de la enfermedad9. El diagnóstico definitivo ha sido realizado por la presencia de hipersensibilidad celular a agentes clastogénicos1,9,10 e confirmado en 4 pacientes por el estudio genético. El FANC-A fue el grupo de complementación más frecuente, lo que esta según referido en la literatura (65% de los pacientes con AF)1. En estos casos, la transmisión hereditaria de la enfermedad es autosómica recesiva. Respecto al tratamiento, solo el transplante de células progenitoras hematopoyéticas es potencialmente curativo de la insuficiencia medular pero se asocia a mortalidad importante-sobrevivencia de 70% a los 2 años cuando se utiliza uno donante hermano HLA-idéntico y 20–40% cuando el donante no está relacionado1. Se recomiendan los andrógenos7,10 que fueran utilizados en todos los casos clínicos presentados, la mayoría con buena respuesta, es decir, aumento del número de células en las tres líneas hematopoyéticas. Habitualmente casi todos los pacientes, después de periodo de tiempo variable, son refractarios a este tratamiento7,10 lo que, hasta el momento, solo se verificó en uno paciente en nuestro estudio. La terapia génica no es una realidad aunque esta sea un área de investigación activa1,6,10. Respecto a la evolución clínica, la morbilidad y mortalidad se relacionan principalmente con fallo medular. Se subraya que estos pacientes tienen un riesgo elevado de neoplasias, sobre todo leucemia mieloide aguda y tumores sólidos. Una de las localizaciones más frecuentes de los tumores sólidos es la cabeza y el cuello3 como se verificó en uno de los pacientes con AF presentados.
La DC es más rara que la AF, estando descritos en la literatura 425 casos2.
Las manifestaciones de la enfermedad surgen en tejidos que están en constante renovación, en general antes de los 10 años de edad1,8. Sin embargo, en lo que respecta la edad de inicio y gravedad de las manifestaciones mucocutáneas existe mucha heterogeneidad clínica, mismo dentro de una misma familia. La estenosis del conducto lagrimal presente en uno paciente con DC es una de varias alteraciones somáticas que se pueden encontrar, como por ejemplo, fibrosis pulmonar y hepática, osteoporosis, estenosis esofágica y estenosis uretral5,8. El diagnóstico de la insuficiencia medular puede solo ser efectuado en edad adulta, pero habitualmente, se desarrolla antes de los 20 años1 como en los 3 casos presentados. El abordaje clínico de estos pacientes es semejante a la de AF. Adicionalmente, se puede efectuar el estudio del largo de los telomeros en leucocitos utilizando citometría de flujo con hibridación in situ fluorescente8. En los 3 pacientes con DC se realizó estudio genético el cual identificó en dos hermanos la misma mutación en el gen DKC1. Este gen que codifica la disquerina es el que más frecuentemente aparece (cerca de 36% de los casos) y se asocia a la forma de transmisión hereditaria recesiva ligada al X5,8. Este hecho explica la mayor prevalencia de la enfermedad en el sexo masculino y también en los casos clínicos de DC presentados. El tratamiento de la insuficiencia medular es semejante a la de AF8. Sin embargo, en la DC, el transplante de progenitores hematopoyéticos se asocia con frecuencia a complicaciones pulmonares/vasculares y, por tanto, tiene una menor tasa de éxito4,8. Respecto a la evolución clínica, tal como en la AF, la mortalidad e morbilidad se relacionan con fallo medular y también existe una predisposición aumentada para neoplasias1,5,8.
De una forma general, las aplasias medulares congénitas son enfermedades graves que se asocian a morbilidad y mortalidad precoz importantes. Se subraya que deben ser consideradas en el diagnóstico diferencial de anemia aplásica, mielodisplasia y leucemia mieloide aguda en niños y jóvenes adultos y, también de tumores sólidos en jóvenes adultos. Los consejos genéticos son fundamentales para el paciente y su familia. El transplante de células progenitoras hematopoyéticas cura la enfermedad medular, pero no modifica las anomalías congénitas ni el riesgo de neoplasias por lo que es fundamental, en estos pacientes, vigilar regularmente su aparición.
Presentación previaReunión anual del Departamento de Hematología y Oncología de la Sociedad Portuguesa de Pediatría que se celebró en Lisboa el 10 de mayo de 2008.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
