





Editado por: Fernando Santos-Simarro. Molecular Diagnostic and Clinical Genetics Unit University Hospital Son Espases. Palma de Mallorca. España
Última actualización: Septiembre 2025
Más datosLa secuenciación de nueva generación (NGS) abarca una gama de tecnologías que han transformado la investigación genómica desde los años 2000. Al permitir la secuenciación de grandes fragmentos de ADN a un costo significativamente menor que la secuenciación de Sanger, la NGS se ha convertido en una herramienta indispensable en los laboratorios moleculares, particularmente en el campo de la genética molecular. Su alta eficiencia y rapidez la convierten en una tecnología de primera línea en el análisis genético.
Un paso crucial para lograr un diagnóstico es el análisis bioinformático. La tecnología de secuenciación de lectura corta genera datos en bruto que deben ser procesados para extraer información significativa e interpretable. Este proceso permite la identificación de vínculos causales entre los hallazgos genéticos y los rasgos fenotípicos. Expertos en Bioinformática Clínica realizan este análisis utilizando herramientas especializadas y pipelines, que tienen en cuenta las características específicas de las plataformas de secuenciación, los protocolos y las enfermedades particulares que se están estudiando.
La revisión de calidad es un complemento esencial del análisis de las pipelines. Su objetivo principal es evaluar qué muestras son adecuadas para el diagnóstico y, en casos donde los resultados son negativos, identificar las razones, ya sean relacionadas con la incidencia u otros factores. Además, la revisión de calidad ofrece información sobre la efectividad general de los procedimientos experimentales.
A pesar de sus muchas ventajas, la NGS aún enfrenta varios desafíos, como la necesidad de tecnologías más eficientes, marcos regulatorios mejorados y una mejor capacitación para el personal médico.
Next Generation Sequencing (NGS) encompasses a range of technologies that have transformed genomic research since the 2000s. By allowing the sequencing of large DNA fragments at a significantly lower cost than Sanger sequencing, NGS has become an indispensable tool in molecular laboratories, particularly in the field of molecular genetics. Its high efficiency and speed make it a first-line technique in genetic analysis.
A crucial step in achieving a diagnosis is bioinformatics analysis. Short-read sequencing technology generates raw data that must be processed to extract meaningful and interpretable information. This process enables the identification of causal links between genetic findings and phenotypic traits. Clinical bioinformatics specialists carry out this analysis using specialized tools and pipelines, which take into account the specific characteristics of the sequencing platforms, protocols and the particular diseases under study.
The quality review is an essential complement to the pipeline analysis. Its primary objective is to assess which samples are suitable for diagnosis and, in cases where results are negative, to identify the reasons, whether they are related to the incidence or other factors. Additionally, the quality review offers insight into the overall effectiveness of the experimental procedures.
Despite its many advantages, NGS still faces several challenges, including the need for more efficient technologies, enhanced regulatory frameworks and improved training of medical staff.
Next Generation Sequencing (NGS) es un término que engloba diversas tecnologías para secuenciar ADN1, revolucionando la investigación genómica desde los años 2000, al permitir la resolución de grandes regiones de ADN a menor coste y tiempo que la secuenciación Sanger. La NGS se ha convertido en una herramienta esencial en los laboratorios moleculares, siendo una tecnología de primera línea por su alto rendimiento y rapidez.
A día de hoy la NGS se emplea en diversos contextos clínicos, aunque con una implantación desigual. Además de su uso en Genética Molecular, está presente en Oncología, Microbiología, Inmunología, Hematología y Salud Pública.
Las plataformas de NGS se dividen en dos grandes categorías: las de lectura corta, que secuencian fragmentos de 100 a 300 pares de bases2, y las de última generación3 (o lectura larga), que pueden leer fragmentos de hasta 100.000 pares de bases, como las de Pacific Biosciences y Oxford Nanopore Technologies. En el entorno clínico, las plataformas Illumina, de lectura corta, son las más utilizadas y sus workflows se consideran un estándar de facto.
En Genética Molecular, los datos de secuenciación de lectura corta deben ser procesados para convertirlos en información útil e interpretable, permitiendo identificar relaciones causales entre hallazgos y fenotipos, y facilitando el manejo clínico del paciente cuando se identifican resultados clínicamente accionables. El análisis de los datos debe ser realizado por especialistas en Bioinformática Clínica con herramientas específicas, considerando las particularidades de las plataformas, los protocolos de secuenciación y las patologías estudiadas. Este análisis se realiza mediante plataformas de análisis o pipelines.
En los últimos años, diversos consorcios han aportado información y recursos valiosos que han permitido conocer en mayor profundidad el perfil de las variaciones, tanto de individuos sanos4 como de pacientes5, mejorando la interpretabilidad de los resultados obtenidos con la NGS.
Sin embargo, la secuenciación masiva, dentro del ámbito diagnóstico, se enfrenta a diversos desafíos6,7 técnicos y metodológicos, como la necesidad de plataformas de secuenciación más eficientes, algoritmos bioinformáticos optimizados, mejoras en la regulación y control de calidad, así como una mayor capacitación del personal médico.
Los próximos apartados abordarán estos aspectos para proporcionar una comprensión completa de la NGS y el estatus actual de otras tecnologías ómicas con fines diagnósticos.
Las tecnologías ómicas y su introducción en la práctica clínicaLas tecnologías ómicas son un conjunto de técnicas que se utilizan para estudiar de manera integral todos los agentes que participan en los procesos moleculares de los seres vivos.
Todas comparten la característica de que generan un volumen masivo de datos que debe analizarse mediante metodologías complejas de base estadística. Además, requieren infraestructuras con gran capacidad de cómputo, recursos para el almacenamiento y gestión de la información, y personal especializado para su análisis y gobernanza.
En el contexto diagnóstico, las ómicas más comunes son la Genómica, la Transcriptómica, la Proteómica, la Metabolómica y la Epigenómica.
De todas ellas, son las tecnologías genómicas (las que analizan el ADN de los individuos, como la NGS) las que tienen una mayor presencia en la rutina clínica. Respecto al resto, en la mayoría de los casos su uso se circunscribe a proyectos de investigación que buscan como último fin su implantación en la clínica. Más adelante se detallan algunos ejemplos de estos usos.
La secuenciación masiva como técnica diagnósticaLa NGS tiene un estatus de test diagnóstico, y en la última década las pruebas moleculares basadas en esta tecnología se han incorporado en la cartera de servicios del sistema de salud español.
Actualmente, la implementación de la NGS sigue siendo desigual entre las comunidades autónomas, afectada por la necesidad de recursos tecnológicos y de personal bioinformático para garantizar la validez de los resultados.
Su introducción en laboratorios moleculares ha marcado un cambio de paradigma: antes, el personal técnico realizaba los experimentos y el facultativo emitía el informe. Ahora, la NGS exige un análisis de datos previo a la interpretación clínica, lo que obliga a los laboratorios a adaptarse a estas nuevas demandas.
Las principales particularidades que presenta la NGS como técnica diagnóstica se pueden resumir en: 1)su deslocalización, y 2)ausencia de puntos de autocontrol.
La deslocalización implica la ejecución de múltiples etapas de manera secuencial para obtener un resultado interpretable a nivel diagnóstico, pudiendo ser realizadas por diferentes laboratorios o instituciones. Cada etapa demanda espacios específicos, recursos especializados y personal experto.
Este flujo de trabajo (fig. 1) comienza cuando el médico solicita la prueba (previa firma del consentimiento informado), continúa con la extracción de ADN del tejido de interés y la creación de librerías de fragmentos de ADN mediante protocolos estandarizados, seguido de la secuenciación en una plataforma de ultrasecuenciación, y culmina con el análisis bioinformático.
 solicita una prueba de NGS, previa firma del consentimiento informado (fase1). Posteriormente se realiza el proceso del test de manera secuencial (fase2) para que, finalmente, los resultados del informe molecular sean comunicados en consulta, y, si es necesario, se ofrecerá asesoramiento genético a los familiares (fase3).")
Diagrama del flujo de trabajo en un test de NGS.
Se muestran las fases necesarias para realizar un test de NGS: inicialmente el paciente acude a consulta y el médico peticionario (facultativo especialista o genetista clínico) solicita una prueba de NGS, previa firma del consentimiento informado (fase1). Posteriormente se realiza el proceso del test de manera secuencial (fase2) para que, finalmente, los resultados del informe molecular sean comunicados en consulta, y, si es necesario, se ofrecerá asesoramiento genético a los familiares (fase3).
Posteriormente, el personal facultativo elabora un informe molecular donde se incluyen los hallazgos (positivos, negativos o no concluyentes) e información relevante sobre el test realizado.
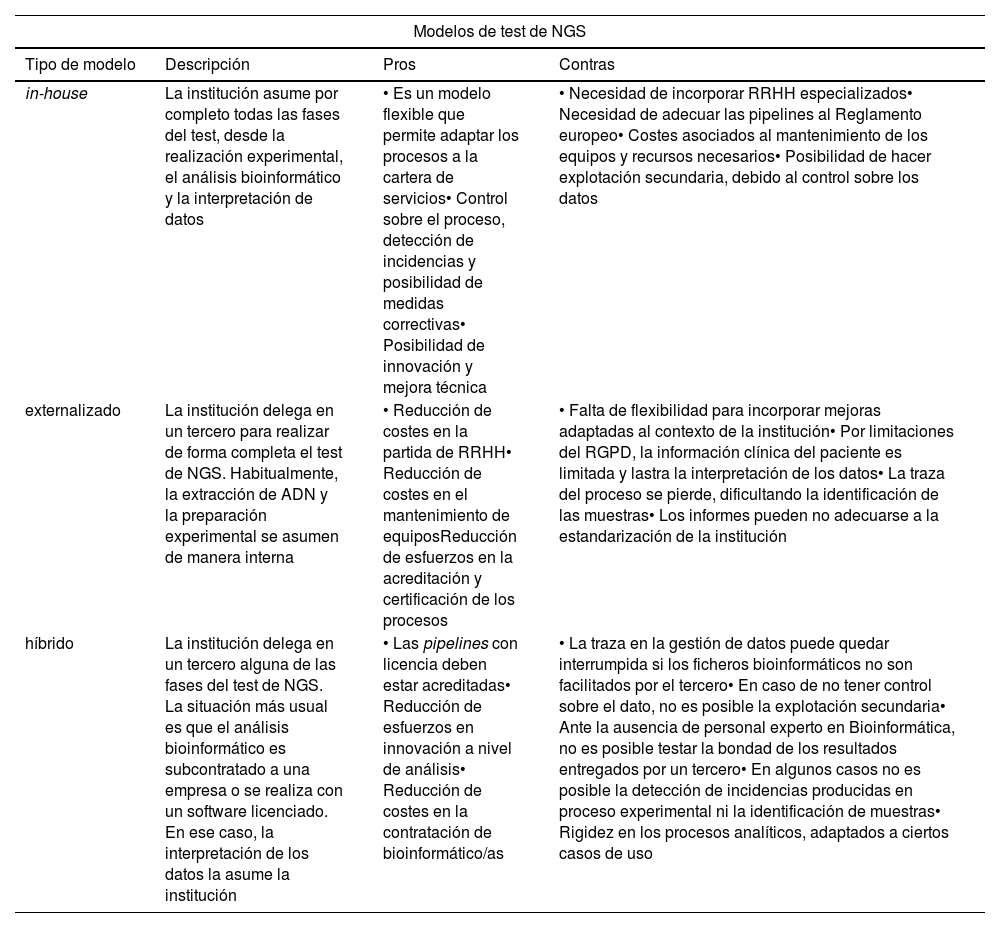
La deslocalización permite que el test, o algunas de sus fases, puedan externalizarse, resultando en modelos de trabajo a)in-house; b)externalizados, o c)híbridos (tabla 1).
Modelos de implantación en el test de NGS
| Modelos de test de NGS | |||
|---|---|---|---|
| Tipo de modelo | Descripción | Pros | Contras |
| in-house | La institución asume por completo todas las fases del test, desde la realización experimental, el análisis bioinformático y la interpretación de datos | • Es un modelo flexible que permite adaptar los procesos a la cartera de servicios• Control sobre el proceso, detección de incidencias y posibilidad de medidas correctivas• Posibilidad de innovación y mejora técnica | • Necesidad de incorporar RRHH especializados• Necesidad de adecuar las pipelines al Reglamento europeo• Costes asociados al mantenimiento de los equipos y recursos necesarios• Posibilidad de hacer explotación secundaria, debido al control sobre los datos |
| externalizado | La institución delega en un tercero para realizar de forma completa el test de NGS. Habitualmente, la extracción de ADN y la preparación experimental se asumen de manera interna | • Reducción de costes en la partida de RRHH• Reducción de costes en el mantenimiento de equiposReducción de esfuerzos en la acreditación y certificación de los procesos | • Falta de flexibilidad para incorporar mejoras adaptadas al contexto de la institución• Por limitaciones del RGPD, la información clínica del paciente es limitada y lastra la interpretación de los datos• La traza del proceso se pierde, dificultando la identificación de las muestras• Los informes pueden no adecuarse a la estandarización de la institución |
| híbrido | La institución delega en un tercero alguna de las fases del test de NGS. La situación más usual es que el análisis bioinformático es subcontratado a una empresa o se realiza con un software licenciado. En ese caso, la interpretación de los datos la asume la institución | • Las pipelines con licencia deben estar acreditadas• Reducción de esfuerzos en innovación a nivel de análisis• Reducción de costes en la contratación de bioinformático/as | • La traza en la gestión de datos puede quedar interrumpida si los ficheros bioinformáticos no son facilitados por el tercero• En caso de no tener control sobre el dato, no es posible la explotación secundaria• Ante la ausencia de personal experto en Bioinformática, no es posible testar la bondad de los resultados entregados por un tercero• En algunos casos no es posible la detección de incidencias producidas en proceso experimental ni la identificación de muestras• Rigidez en los procesos analíticos, adaptados a ciertos casos de uso |
Actualmente, en entornos hospitalarios, existen tres modelos diferentes de test de NGS: in-house (donde la institución asume todos los pasos del test), externalizado (donde se subcontrata el servicio a un tercero) o híbrido (cuando alguna de las fases del test se delega en un tercero). En la tabla se lista los aspectos a favor y en contra de cada uno de los modelos.
La fragmentación de subprocesos requiere protocolos de comunicación normalizados entre todas las partes que aseguren la trazabilidad y la correcta gestión del proceso diagnóstico. La supervisión y la monitorización del proceso son esenciales para garantizar la validez analítica y clínica del test8, incluso en modelos externalizados o híbridos.
La validez global del test depende fuertemente del proceso analítico, ya que es en el proceso bioinformático donde se van a detectar el grueso de incidencias, como cambios de muestra, contaminaciones, muestras de baja calidad, etc., y se van a identificar finalmente qué muestras son aptas para el diagnóstico.
Sin embargo, uno de los desafíos de la NGS es la falta de puntos de autocontrol durante el proceso, más allá de los existentes en las plataformas de secuenciación. Esto es crítico, especialmente en la determinación de variantes, donde pipelines mal calibradas o con salidas primarias del secuenciador incompletas pueden generar archivos con información deficiente.
Además, la falta de buenas prácticas estandarizadas y la deslocalización entre laboratorios que participan de una misma prueba dificultan la implementación de medidas correctivas que aseguren la calidad y la integridad del proceso. Esto resalta la importancia de adoptar marcos de calidad por parte de los laboratorios clínicos, como las normas ISO, para garantizar la seguridad del paciente. Estas cuestiones se abordarán más adelante.
Otro aspecto importante de la NGS es la posibilidad de realizar estudios dirigidos mediante paneles de genes asociados a enfermedades, enfocándose en regiones específicas del genoma reduciendo costes y tiempo de análisis. Así mismo, cuando se requieren análisis más amplios, es posible secuenciar el exoma completo (WES) o el genoma completo (WGS).
Finalmente, debemos destacar la necesidad de gestión de un gran volumen de datos que se generan en cada test realizado. El tamaño por prueba varía en función de múltiples factores, como la extensión de la región secuenciada, el grado de amplificación (llamado profundidad de lecturas) y el número de muestras que se secuencian de manera conjunta.
Como referencia, se pueden consultar las especificaciones de la plataforma NovaSeq6000 de Illumina9, que ofrece una visión general sobre la escala de los tamaños de archivo. La salida bruta del secuenciador varía entre 230gigabyte (GB) y 3terabyte (TB), mientras que un estudio completo, considerando todos los archivos generados tanto en la secuenciación como en el análisis, puede ocupar entre 560GB y 6,5TB.
Los datos de NGS deben ser tratados como datos médicos, al igual que la imagen médica u otra información clínica. Bajo esta perspectiva, y conforme a la Ley 41/2002 sobre la autonomía del paciente, su conservación debe garantizarse por un mínimo de 5años.
Por ello, contar con una infraestructura de almacenamiento adecuada es fundamental, y requiere una planificación eficiente que asegure la custodia de los datos genómicos a medio plazo.
Otras ómicas aplicadas al diagnósticoAdemás de la genómica, otras tecnologías ómicas están ganando relevancia en el ámbito clínico. Aunque ya se utilizan para el diagnóstico en algunos casos, su aplicación sigue siendo mayoritariamente preclínica.
Estas tecnologías juegan un papel clave en el descubrimiento, el análisis y la validación de nuevas terapias, biomarcadores y herramientas diagnósticas, fundamentales para el avance de la medicina personalizada.
La proteómica, que analiza a gran escala la presencia de proteínas en una muestra, ha experimentado avances significativos en los últimos años gracias a mejoras técnicas y análisis bioinformáticos más efectivos10. Un ejemplo destacado es el test Overa, desarrollado por Vermillion, Inc., que utiliza espectrometría de masas en tándem (LC-MS/MS) para evaluar el riesgo de cáncer de ovario en mujeres con masas pélvicas. Este test recibió la aprobación de la FDA en 2016.
El estudio de metabolitos es conocido en medicina pediátrica, especialmente en el cribado neonatal de errores congénitos del metabolismo. La metabolómica representa un avance relevante al permitir el análisis no dirigido de miles de metabolitos, facilitando la identificación de firmas metabólicas en fluidos y tejidos. Actualmente se investiga su aplicación en diversas patologías, como la diabetes tipo2, donde la detección temprana podría mejorar el pronóstico de los pacientes11.
Otro ámbito de interés es la oncología de precisión, donde es clave poder establecer biomarcadores para la detección precoz, la clasificación tumoral y la respuesta a tratamientos. En este sentido, el análisis del epigenoma está aportando casos de éxito como el uso de mutaciones activadoras de EGFR como biomarcadores, lo que ha mejorado significativamente la supervivencia de los pacientes con cáncer de pulmón de células no pequeñas (NSCLC)12.
En el ámbito de la Genética Molecular hay que destacar las metodologías de integración (llamadas también «multiómicas»), que permiten fusionar información para mejorar el diagnóstico de pacientes cuyo mecanismo causal no ha sido identificado a pesar de mostrar fenotipos característicos.
Así, la inclusión de muestras de RNA-Seq en estudios moleculares permite analizar el transcriptoma del paciente, facilitando la identificación de alteraciones en la maquinaria de splicing y la detección de genes con expresión aberrante13.
El estudio de la firma epigenética14 está mostrando también su utilidad en pacientes con enfermedades del neurodesarrollo, ya que permite la clasificación de pacientes con una manifestación sindrómica compleja o con fenotipos similares, como el síndrome de Kabuki y el síndrome de CHARGE, mejorando la precisión en el diagnóstico.
Análisis de datos de NGS: pipelines bioinformáticasLlamamos pipeline bioinformática al conjunto de herramientas y algoritmos que analizan la salida bruta de las plataformas de secuenciación e identifican las variantes genéticas que porta el paciente. El término técnico pipeline se emplea ampliamente en los laboratorios de diagnóstico, al igual que otros conceptos propios de la Bioinformática que han sido adoptados sin necesidad de traducción; por ello, los usaremos de este modo a lo largo del texto.
Las pipelines suelen integrar herramientas de software libre y código abierto, aunque también existen versiones bajo licencia o que incorporan herramientas comerciales como complemento.
Las pipelines deben estar adaptadas al tipo de variante y su origen (germinal o somático), lo que exige metodologías específicas para cada caso.
En línea germinal, las variantes que usualmente se caracterizan son: 1)variantes puntuales (SNVs, por su acrónimo en inglés), 2)pequeñas inserciones o deleciones (INDELs), 3)cambios en número de copias (CNVs), y 4) otras variantes estructurales (SVs).
La necesidad de especialización y parametrización específica para atender cada caso de uso hace que a menudo se opte por pipelines in-house en lugar de soluciones comerciales. Las pipelines in-house se desarrollan en los propios laboratorios y son más flexibles, pero su uso plantea cuestiones normativas y de calidad, mientras que las comerciales ofrecen otras ventajas y están certificadas para algunos contextos específicos. La tabla 1 resume los pros y los contras de ambos enfoques.
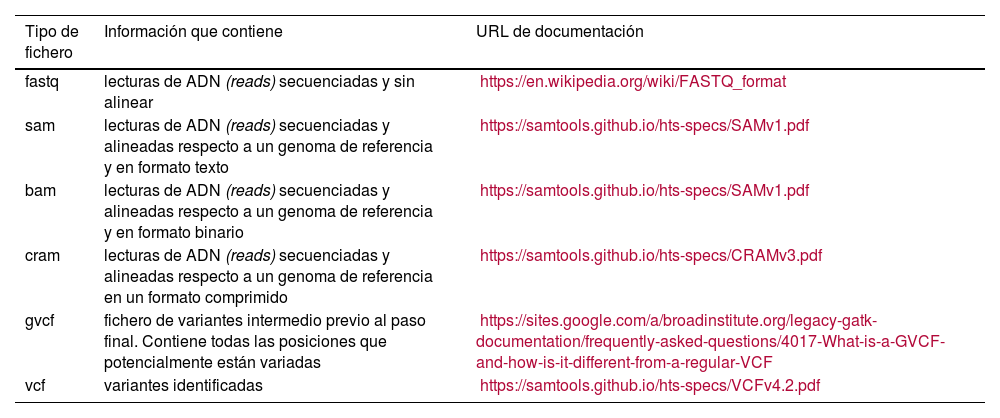
Los siguientes apartados describen la lógica de análisis de una pipeline. Los formatos de archivo generados en cada etapa, junto con las herramientas más utilizadas, se detallan en las tablas 2-5, que también incluyen sus respectivas referencias.
Formatos de ficheros en el proceso de análisis bioinformático
| Tipo de fichero | Información que contiene | URL de documentación |
|---|---|---|
| fastq | lecturas de ADN (reads) secuenciadas y sin alinear | https://en.wikipedia.org/wiki/FASTQ_format |
| sam | lecturas de ADN (reads) secuenciadas y alineadas respecto a un genoma de referencia y en formato texto | https://samtools.github.io/hts-specs/SAMv1.pdf |
| bam | lecturas de ADN (reads) secuenciadas y alineadas respecto a un genoma de referencia y en formato binario | https://samtools.github.io/hts-specs/SAMv1.pdf |
| cram | lecturas de ADN (reads) secuenciadas y alineadas respecto a un genoma de referencia en un formato comprimido | https://samtools.github.io/hts-specs/CRAMv3.pdf |
| gvcf | fichero de variantes intermedio previo al paso final. Contiene todas las posiciones que potencialmente están variadas | https://sites.google.com/a/broadinstitute.org/legacy-gatk-documentation/frequently-asked-questions/4017-What-is-a-GVCF-and-how-is-it-different-from-a-regular-VCF |
| vcf | variantes identificadas | https://samtools.github.io/hts-specs/VCFv4.2.pdf |
Listado de los distintos formatos de ficheros que se crean durante el análisis bioinformático. Se detalla su contenido y una URL donde se podrán encontrar una descripción en profundidad de las especificaciones de cada formato.
Herramientas utilizadas en la primera fase de la pipeline
| Nombre | Paso | URL |
|---|---|---|
| bcl2fastq | demultiplexación Illumina | https://emea.support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html |
| Trimmomatic | Trimming | http://www.usadellab.org/cms/?page=trimmomatic |
| Cutadapt | Trimming | https://cutadapt.readthedocs.io/en/stable/ |
| BBDuk | Trimming | https://github.com/BioInfoTools/BBMap |
| BWA-MEM | Alineamiento | https://github.com/lh3/bwa |
| Bowtie2 | Alineamiento | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Minimap2 | Alineamiento | https://github.com/lh3/minimap2 |
| Picard | Suites generales | https://broadinstitute.github.io/picard/ |
| Samtools | Suites generales | http://www.htslib.org/ |
La fase inicial, o «pasos comunes», consiste en a)el filtrado o trimming; b)el alineamiento, y c)el filtrado de duplicados. Las herramientas más comunes para realizar estas tareas se listan en la tabla junto con la URL donde se podrá encontrar el software de cada herramienta.
Herramientas para la determinación de variantes
| Nombre | Tipo de variante | Tipo de estudio | URL |
|---|---|---|---|
| GATK4/ | SNV/INDELs | panel/WES/WGS | https://gatk.broadinstitute.org/hc/en-us |
| DeepVariant | SNV/INDELs | panel/WES/WGS | https://github.com/google/deepvariant |
| DECON | CNVs | panel/WES | https://github.com/RahmanTeam/DECoN |
| XHMM | CNVs | panel/WES | https://github.com/RRafiee/XHMM |
| panelcn.MOPS | CNVs | panel/WES | https://github.com/bioinf-jku/panelcn.mops |
| ExomeDepth | CNVs | panel/WES | https://github.com/vplagnol/ExomeDepth |
| CODEX2 | CNVs | panel/WES | https://github.com/yuchaojiang/CODEX2 |
| Genome STRiP | SVs | WGS | https://software.broadinstitute.org/software/genomestrip |
| Delly | SVs | WGS | https://github.com/dellytools/delly |
| Manta | SVs | WGS | https://github.com/Illumina/manta |
| LUMPY | SVs | WGS | https://github.com/arq5x/lumpy-sv |
| ExpansionHunter | expansiones de secuencias repetitivas | WES/WGS | https://github.com/Illumina/ExpansionHunter |
| GangSTR | expansiones de secuencias repetitivas | WES/WGS | https://github.com/gymreklab/GangSTR |
Listado de herramientas más conocidas para la determinación de las variantes en los reads alineados. Las variantes que se pueden caracterizar son SNVs, INDELs, CNVs y otras SVs.
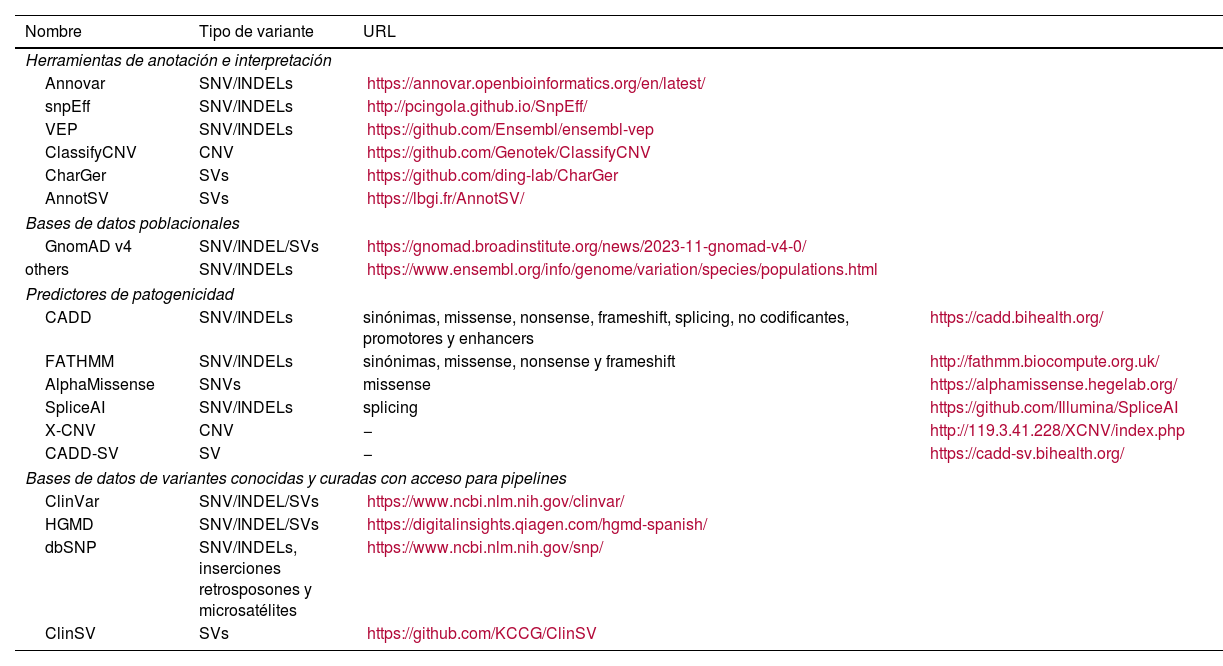
Herramientas de anotación de variantes
| Nombre | Tipo de variante | URL | |
|---|---|---|---|
| Herramientas de anotación e interpretación | |||
| Annovar | SNV/INDELs | https://annovar.openbioinformatics.org/en/latest/ | |
| snpEff | SNV/INDELs | http://pcingola.github.io/SnpEff/ | |
| VEP | SNV/INDELs | https://github.com/Ensembl/ensembl-vep | |
| ClassifyCNV | CNV | https://github.com/Genotek/ClassifyCNV | |
| CharGer | SVs | https://github.com/ding-lab/CharGer | |
| AnnotSV | SVs | https://lbgi.fr/AnnotSV/ | |
| Bases de datos poblacionales | |||
| GnomAD v4 | SNV/INDEL/SVs | https://gnomad.broadinstitute.org/news/2023-11-gnomad-v4-0/ | |
| others | SNV/INDELs | https://www.ensembl.org/info/genome/variation/species/populations.html | |
| Predictores de patogenicidad | |||
| CADD | SNV/INDELs | sinónimas, missense, nonsense, frameshift, splicing, no codificantes, promotores y enhancers | https://cadd.bihealth.org/ |
| FATHMM | SNV/INDELs | sinónimas, missense, nonsense y frameshift | http://fathmm.biocompute.org.uk/ |
| AlphaMissense | SNVs | missense | https://alphamissense.hegelab.org/ |
| SpliceAI | SNV/INDELs | splicing | https://github.com/Illumina/SpliceAI |
| X-CNV | CNV | − | http://119.3.41.228/XCNV/index.php |
| CADD-SV | SV | − | https://cadd-sv.bihealth.org/ |
| Bases de datos de variantes conocidas y curadas con acceso para pipelines | |||
| ClinVar | SNV/INDEL/SVs | https://www.ncbi.nlm.nih.gov/clinvar/ | |
| HGMD | SNV/INDEL/SVs | https://digitalinsights.qiagen.com/hgmd-spanish/ | |
| dbSNP | SNV/INDELs, inserciones retrosposones y microsatélites | https://www.ncbi.nlm.nih.gov/snp/ | |
| ClinSV | SVs | https://github.com/KCCG/ClinSV | |
En la tabla se muestran las herramientas y los recursos más conocidos para realizar este paso. Primero, se listan algunos de los programas de anotación más utilizados con su URL para acceder al software. Después, se indican las bases de datos más conocidas para establecer la frecuencia alélica de las variantes. Además, se incluye un listado de predictores de patogenicidad con sus correspondientes URL. Finalmente, se indican algunas bases de datos de variantes conocidas cuyo efecto está revisado por expertos y que pueden ser accedidas por pipelines bioinformáticas.
El flujo de trabajo de una pipeline se muestra en la figura 2B. Generalmente, existe una etapa inicial que incluye: 1)procesar la salida del secuenciador; 2)filtrar, y 3)alinear las lecturas de ADN contra un genoma de referencia.
 Ejemplo de una variante puntual o SNV en heterocigosis en el gen AGL. Los reads alineados aparecen en gris, y el contenido nucleotídico del fragmento del exón mostrado se puede ver abajo en la figura. B)Etapas del proceso de análisis bioinformático que deben ejecutarse de manera secuencial mediante una pipeline. C)Información asociada a la variante de A) con sus anotaciones correspondientes que permiten la interpretación de la variante.")
Resumen del análisis bioinformático.
A) Ejemplo de una variante puntual o SNV en heterocigosis en el gen AGL. Los reads alineados aparecen en gris, y el contenido nucleotídico del fragmento del exón mostrado se puede ver abajo en la figura. B)Etapas del proceso de análisis bioinformático que deben ejecutarse de manera secuencial mediante una pipeline. C)Información asociada a la variante de A) con sus anotaciones correspondientes que permiten la interpretación de la variante.
En las plataformas de secuenciación comercializadas por Illumina es necesario realizar un proceso de demultiplexado sobre la salida primaria del secuenciador. Este proceso consiste en separar los datos brutos generados por el secuenciador en diferentes muestras individuales, asignando a cada una sus correspondientes archivos .fastq, que contienen las lecturas de ADN (en adelante, reads). En caso de que la secuenciación sea paired-end, se obtienen lecturas desde ambos extremos de cada fragmento de ADN, lo que genera dos archivos .fastq por muestra.
Luego, se aplica un filtrado (trimming) para eliminar reads de baja calidad, artefactos y secuencias adaptadoras. Posteriormente, cada read filtrado debe alinearse con respecto a un genoma de referencia, cuya elección influye en la caracterización de variantes15, por lo que es un aspecto importante a considerar.
Las lecturas alineadas se almacenan en archivos .bam, que pueden visualizarse en visores genómicos. La inspección manual de regiones de interés proporciona información valiosa a los expertos.
Cada read tiene un valor de calidad determinado por varios factores. Por ejemplo, en secuenciación de lectura corta las regiones genómicas poco complejas y repetitivas suelen estar cubiertas por alineamientos de baja calidad, lo que puede sesgar los conteos de cobertura y dificultar la identificación de variantes.
Determinación de variantesTras asignar coordenadas genómicas a cada lectura de ADN, se identifican las variantes genéticas en un proceso denominado «llamada de variantes». Este paso es altamente especializado, ya que los algoritmos empleados varían según la alteración a detectar.
Estos algoritmos se basan en métodos estadísticos y requieren una profundidad de lectura adecuada para identificar patrones. La figura 2A muestra una variante puntual (SNV) en el gen AGL.
Además de identificar la variante, es esencial determinar su cigosidad, es decir, si está en heterocigosis (un solo alelo mutado) o en homocigosis (ambos alelos mutados). Sin embargo, el proceso experimental habitualmente elimina el origen alélico de cada lectura, por lo que esta información debe inferirse posteriormente.
La información asociada a las variantes conlleva cierto grado de incertidumbre, y existe la posibilidad de que algunas sean falsos positivos. Por ello, su fiabilidad debe evaluarse a través de los scores de las herramientas disponibles, teniendo en cuenta los sesgos presentes en determinadas regiones genómicas. Además, se recomienda visualizar las variantes en su contexto genómico para una interpretación más precisa.
Los algoritmos para SNVs han evolucionado en la última década, y actualmente se analizan las muestras en grupos en lugar de cada una individualmente para minimizar artefactos recurrentes en lotes secuenciados conjuntamente.
GATK416 es la herramienta más conocida para este enfoque, mientras que DeepVariant17, basada en redes neuronales, se adapta a distintos contextos, incluido el somático. Barbitoff (2022)18 ofrece una revisión general sobre herramientas bioinformáticas para SNVs e INDELs.
Para detectar cambios en el número de copias (CNVs), el método más común estima la dosis génica mediante la profundidad de lectura normalizada frente a una población control, un enfoque frecuente en estudios de captura dirigida. En WGS se combinan diversas estrategias para identificar CNVs y variantes estructurales (SVs), como grandes deleciones, duplicaciones, inversiones y translocaciones19.
Otro tipo de variante de interés, especialmente en enfermedades neurodegenerativas hereditarias, son las repeticiones en tándem y tripletes repetidos. Para conocer mejor su detección por NGS se recomienda la revisión de Genomics England20.
A diferencia de SNVs/INDELs, la detección de SVs con lecturas cortas se usa principalmente con fines de cribado debido a la alta sensibilidad y la baja especificidad de los algoritmos, lo que genera una elevada tasa de falsos positivos. Esta limitación surge por restricciones tecnológicas y diseños que secuencian solo exones para maximizar la rentabilidad. En entornos clínicos, se exploran enfoques híbridos que combinan múltiples tecnologías con resultados prometedores en investigación.
Se recomienda validar las CNVs y SVs detectadas con lectura corta mediante pruebas independientes. Sin embargo, esto no siempre es posible debido a la falta de sondas comerciales para MLPA (MRC Holland) o arrays CGH/SNPs que cubran las regiones de interés. Por ello, es crucial un estudio previo para definir qué regiones incluir en los test de NGS, excluyendo aquellas sin un método de validación claro.
AnotaciónEn este paso, las variantes se enriquecen con información (esto es, anotaciones) para su interpretación en un contexto funcional, poblacional y clínico.
Para describir SNVs e INDELs se debe utilizar la nomenclatura estándar HGVS21, tomando como referencia un transcrito seleccionado cuidadosamente22,23. El efecto de la variante depende del transcrito elegido y podría afectar la precisión del diagnóstico.
La información funcional debe respaldarse con predicciones in silico mediante programas que estimen la probabilidad de que un SNV/INDEL sea deletéreo24. La tabla 5 incluye un listado de los predictores más utilizados.
La información poblacional permite determinar la presencia de variantes en la población de referencia. La base de datos gnomAD25 es uno de los recursos más completos, ya que contiene datos de 195.000 individuos considerados población control.
También debe integrarse información de variantes conocidas y revisadas por expertos, como la que aporta dbSNP, ClinVar y HGMD (esta última requiere licencia).
Aunque el flujo de trabajo para SNVs e INDELs está estandarizado, las herramientas para la anotación de CNVs y SVs son aún limitadas y carecen de estándares ampliamente aceptados. Es recomendable considerar herramientas como ClassifyCNV y AnnotSV, así como CharGer, que incorpora los criterios del American College of Medical Genetics and Genomics (ACMG)26, reconocidos como referencia para la clasificación de variantes.
Filtrado y priorización e interpretaciónLa falta de una definición clara de las funciones de los bioinformáticos clínicos genera diferentes perspectivas sobre quién debería encargarse del filtrado y de la priorización de variantes.
Es recomendable que este paso sea realizado por expertos moleculares, quienes deben determinar la patogenicidad de las variantes candidatas siguiendo el sistema de clasificación ACMG.
Sin embargo, en estudios genéticos complejos se requiere de la panelización virtual y/o filtros avanzados mediante algoritmos específicos. Por tanto, es fundamental que los bioinformáticos colaboren estrechamente con los genetistas moleculares para apoyar la toma de decisiones en la fase final del test de NGS.
Existen diversas herramientas de filtrado y priorización que facilitan estas tareas; la mayoría requieren licencia, permitiendo manejar amplias listas de variantes, aunque su descripción queda fuera del alcance de esta revisión.
Otras consideracionesLas metodologías de análisis evolucionan constantemente, por lo que es fundamental actualizar las pipelines con algoritmos más eficaces y bases de datos actualizadas. Dado que no existe una normativa que regule la frecuencia de estas actualizaciones, es crucial documentar detalladamente las versiones de las pipelines, el software utilizado y los recursos empleados, asegurando que esta información se recoja en los informes moleculares para trazar el proceso analítico.
Las pipelines necesitan ser monitorizadas para conocer su rendimiento a lo largo del tiempo y asegurar su validez ante cambios introducidos en el proceso diagnóstico. También, en pipelines in-house, en la fase de desarrollo previo es fundamental ajustar correctamente los parámetros de los programas.
Por ello, es necesario utilizar muestras de referencia bien caracterizadas, como las del consorcio Genome in a Bottle (GIAB)27, con el fin de optimizar el rendimiento y evaluar la reproducibilidad de los resultados. Sin embargo, no están disponibles materiales de referencia para ciertas alteraciones, lo que lastra la implantación de algoritmos bien calibrados en la rutina diagnóstica.
En el caso de la muestra NA12878/HG001 de GIAB, se pueden adquirir viales con ADN de este individuo, lo que permite validar tanto el proceso bioinformático de forma independiente como el flujo completo, desde las primeras etapas experimentales.
Existen, además, evaluaciones periódicas para establecer un rendimiento estándar de referencia, como los esquemas de evaluación de EMQN. Es altamente recomendable seguir alguna evaluación externa que permita evaluar la competencia de las pipelines.
La reproducibilidad de los resultados obtenidos con las pipelines puede verse afectada por la infraestructura hardware y las versiones de las herramientas empleadas. Por este motivo, se aconseja ejecutar las pipelines en contenedores28, ya que estos encapsulan el código con todas las dependencias y archivos necesarios, garantizando la consistencia en cualquier entorno y mejorando la portabilidad, la traza y la reproducibilidad.
Esquema de análisis de calidadLa pipeline de análisis debe complementarse con procesos de revisión de calidad. Su objetivo principal es determinar qué muestras son aptas para el diagnóstico y, en los casos negativos, objetivar el motivo, ya sea por incidencia u otras razones. Además, proporcionar información sobre la efectividad del proceso experimental.
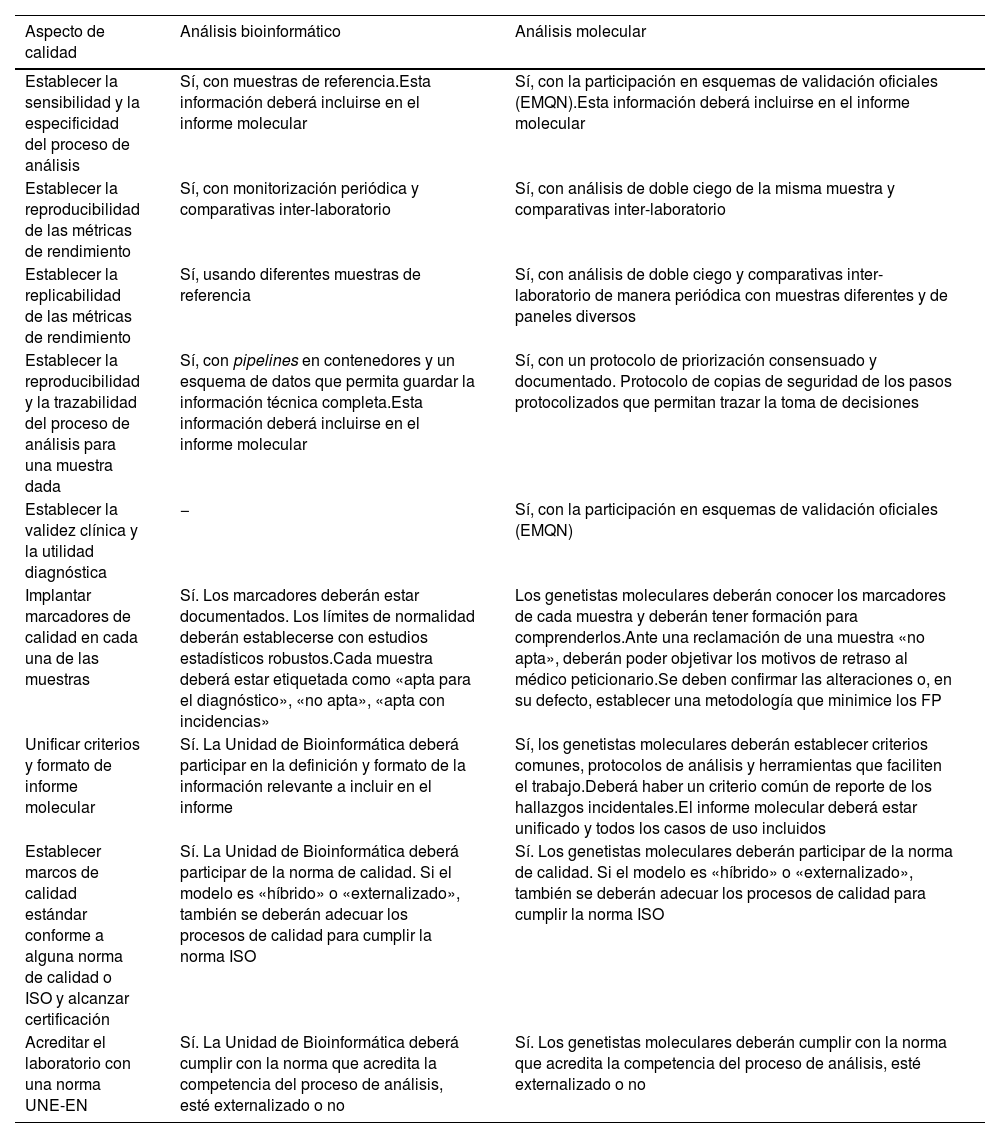
Es importante señalar que los marcadores de calidad no están estandarizados y, hasta el momento, no existe un consenso sobre cuáles deberían ser analizados e incluidos en los informes de forma obligatoria. La descripción detallada de los marcadores más comúnmente utilizados excede el alcance de este artículo, ya que requiere un tratamiento más amplio y exhaustivo. No obstante, la tabla 6 presenta una selección de aspectos de calidad que deben considerarse tanto para la supervisión del proceso como para la revisión de las muestras.
Aspectos de calidad para supervisar el test de NGS y analizar la idoneidad de las muestras
| Aspecto de calidad | Análisis bioinformático | Análisis molecular |
|---|---|---|
| Establecer la sensibilidad y la especificidad del proceso de análisis | Sí, con muestras de referencia.Esta información deberá incluirse en el informe molecular | Sí, con la participación en esquemas de validación oficiales (EMQN).Esta información deberá incluirse en el informe molecular |
| Establecer la reproducibilidad de las métricas de rendimiento | Sí, con monitorización periódica y comparativas inter-laboratorio | Sí, con análisis de doble ciego de la misma muestra y comparativas inter-laboratorio |
| Establecer la replicabilidad de las métricas de rendimiento | Sí, usando diferentes muestras de referencia | Sí, con análisis de doble ciego y comparativas inter-laboratorio de manera periódica con muestras diferentes y de paneles diversos |
| Establecer la reproducibilidad y la trazabilidad del proceso de análisis para una muestra dada | Sí, con pipelines en contenedores y un esquema de datos que permita guardar la información técnica completa.Esta información deberá incluirse en el informe molecular | Sí, con un protocolo de priorización consensuado y documentado. Protocolo de copias de seguridad de los pasos protocolizados que permitan trazar la toma de decisiones |
| Establecer la validez clínica y la utilidad diagnóstica | − | Sí, con la participación en esquemas de validación oficiales (EMQN) |
| Implantar marcadores de calidad en cada una de las muestras | Sí. Los marcadores deberán estar documentados. Los límites de normalidad deberán establecerse con estudios estadísticos robustos.Cada muestra deberá estar etiquetada como «apta para el diagnóstico», «no apta», «apta con incidencias» | Los genetistas moleculares deberán conocer los marcadores de cada muestra y deberán tener formación para comprenderlos.Ante una reclamación de una muestra «no apta», deberán poder objetivar los motivos de retraso al médico peticionario.Se deben confirmar las alteraciones o, en su defecto, establecer una metodología que minimice los FP |
| Unificar criterios y formato de informe molecular | Sí. La Unidad de Bioinformática deberá participar en la definición y formato de la información relevante a incluir en el informe | Sí, los genetistas moleculares deberán establecer criterios comunes, protocolos de análisis y herramientas que faciliten el trabajo.Deberá haber un criterio común de reporte de los hallazgos incidentales.El informe molecular deberá estar unificado y todos los casos de uso incluidos |
| Establecer marcos de calidad estándar conforme a alguna norma de calidad o ISO y alcanzar certificación | Sí. La Unidad de Bioinformática deberá participar de la norma de calidad. Si el modelo es «híbrido» o «externalizado», también se deberán adecuar los procesos de calidad para cumplir la norma ISO | Sí. Los genetistas moleculares deberán participar de la norma de calidad. Si el modelo es «híbrido» o «externalizado», también se deberán adecuar los procesos de calidad para cumplir la norma ISO |
| Acreditar el laboratorio con una norma UNE-EN | Sí. La Unidad de Bioinformática deberá cumplir con la norma que acredita la competencia del proceso de análisis, esté externalizado o no | Sí. Los genetistas moleculares deberán cumplir con la norma que acredita la competencia del proceso de análisis, esté externalizado o no |
Listado de aspectos a tener en cuenta en un proceso de calidad tanto en la etapa de análisis bioinformático como la posterior interpretación de variantes en la fase de análisis molecular y redacción de informe al paciente.
La secuenciación de lectura corta presenta limitaciones debido al tamaño reducido de los fragmentos, lo que resulta en alineamientos ambiguos en ciertas regiones. En especial, fragmentos de baja complejidad (homopolímeros), regiones repetitivas (repeticiones en tándem) o con alta homología (duplicaciones segmentales y pseudogenes). Esto dificulta determinar con precisión la localización de los reads, especialmente cuando las regiones repetidas superan el tamaño del fragmento.
Los algoritmos de alineamiento asignan múltiples localizaciones genómicas a cada read, reduciendo la calidad y sesgando la identificación de variantes, produciendo variantes artefactuales o una falta en su identificación. Un ejemplo es el gen PKD1, crucial para el diagnóstico de poliquistosis renal, donde la presencia de pseudogenes reduce drásticamente las variantes identificadas, requiriendo métodos más avanzados29.
Nuevos protocolos, como linked reads y tecnologías de lectura larga, están demostrando su efectividad en la resolución de ciertas regiones que suponen un reto tecnológico, como las que codifican los antígenos leucocitarios humanos (HLA)30.
Romper la barrera diagnóstica implica explorar nuevas aproximaciones e incorporar otras tecnologías ómicas, tal y como se ha expuesto en apartados anteriores.
Explotación de datos de NGSLos datos genómicos deben tratarse como datos médicos y, por tanto, deben ser gestionados por los mismos responsables que custodian la historia clínica en los sistemas de salud.
El reanálisis por ajustes en la sospecha diagnóstica, nuevos familiares afectados o la explotación del historial de variantes exige que los datos sigan accesibles. Por ello, los sistemas de archivado deben garantizar su consulta durante el período legal y más allá de este.
Estructurar el histórico de pacientes facilita la interpretación de variantes candidatas, ya que su presencia/ausencia en la cohorte permite establecer sus clasificaciones con más confianza. Las consultas y la interconexión entre diferentes bases de datos pueden realizarse mediante el protocolo Beacon31 y permiten incorporar información clínica del paciente.
La explotación secundaria es fundamental para alimentar el ciclo de investigación-desarrollo-implementación para que los laboratorios ajusten sus prácticas a los avances tecnológicos, siempre y cuando los consentimientos informados permitan la reutilización de la información.
Finalmente, es importante anonimizar los archivos de NGS para cumplir con el Reglamento general de protección de datos (RGPD), evitando incluir información personal de los pacientes.
Aspectos normativosDesde 2014, Eurogentest ha emitido recomendaciones para mejorar la precisión de los test genéticos, como la guía publicada para el análisis de WGS32. En Europa, NEQAS (Reino Unido) y VKGL (Países Bajos) son referencia de buenas prácticas en NGS, ante la ausencia de un marco regulatorio. Sin embargo, la acreditación ISO 15189 está muy extendida en los laboratorios de NGS dentro del ámbito europeo.
Las pipelines in-house son consideradas productos sanitarios para diagnóstico in vitro y están reguladas por el Reglamento (UE) 2017/746. A nivel nacional, se está actualizando la normativa que reemplazará al Real Decreto 1662/2000, y un grupo de trabajo europeo ha elaborado una guía al respecto33.
Si se eligen servicios de un tercero para custodiar datos en la nube, este debe demostrar que cumple con sus obligaciones respecto a la gestión de datos personales y de salud según el RGPD34.
DiscusiónEl análisis bioinformático en pruebas de NGS es complejo y demanda personal especializado, una infraestructura adecuada y capacitación del personal facultativo para interpretar correctamente los resultados.
Es esencial la supervisión y el control de calidad para garantizar la validez analítica y clínica del test. Esto es especialmente relevante en casos en que se delega el análisis bioinformático a un tercero.
Aunque la tecnología de lectura corta ha mostrado gran eficiencia y una elevada tasa diagnóstica, es necesario integrar nuevas tecnologías para abordar los casos sin diagnóstico y romper el techo diagnóstico en aquellos donde las bases genéticas no son conocidas o la manifestación clínica es compleja.
Los aspectos normativos son clave, pero aún falta avanzar hacia prácticas estandarizadas y consensuadas por la comunidad de expertos. En este sentido, las administraciones deben asumir un papel más activo para promover la estandarización y las buenas prácticas en el área.
Declaración sobre el uso de inteligencia artificialDurante la preparación de este trabajo, la autora ha utilizado ChatGPT con el fin de mejorar la legibilidad del texto escrito por ella. Tras utilizar esta herramienta/servicio, la autora ha revisado y editado el contenido según fue necesario y asume plena responsabilidad por el contenido de la publicación.
Conflicto de interesesLa autora declara que no existen conflictos de intereses relacionados con esta publicación.
