










El síndrome STXBP1 es una enfermedad genética que afecta a uno de los mecanismos reguladores de la liberación de neurotransmisores por parte de las vesículas sinápticas, por lo que tiene serias implicaciones para el neurodesarrollo. Suele manifestarse en los primeros días o meses de vida e incluye con mucha frecuencia epilepsia, retraso psicomotor y discapacidad intelectual. Aunque inicialmente se consideró una encefalopatía epiléptica precoz, el aumento de los casos diagnosticados y el avance de la investigación han ido ampliando el fenotipo y caracterizando esta enfermedad como un trastorno del neurodesarrollo. Además de estar asociada a alteraciones epilépticas, esta mutación genética puede estar asociada a muchos casos de discapacidad intelectual y trastornos del movimiento cuya causa se desconoce.
ObjetivosDescribir las características de los pacientes identificados en España con el síndrome STXBP1 y las implicaciones para el diagnóstico de dichas características.
Pacientes y métodosSe presentan los datos de 17 personas diagnosticadas en España de síndrome STXBP1 de edades comprendidas entre los 2 y los 17 años.
ConclusionesExiste un claro infradiagnóstico del síndrome STXBP1 en nuestro país. Además de la diversidad inherente al trastorno, con el aumento del número de diagnósticos la variabilidad fenotípica se amplía aún más. Se hace, por tanto, necesaria la descripción de signos de alarma que permitan identificar a aquellas personas con manifestaciones menos prototípicas de la alteración.
STXBP1 syndrome is a genetic disorder that affects one of the regulatory mechanisms of neurotransmitter release by the synaptic vesicles and has serious implications for neurodevelopment. Symptoms usually appear in the first days or months of life, and very often include epilepsy, psychomotor delay, and intellectual disability. Although it was initially regarded as an early epileptic encephalopathy, the increase in the number of cases diagnosed, as well as the advances in research have been expanding the phenotype and characterising this disease as a disorder of neurodevelopment. Furthermore, on being linked to epileptic problems, this genetic mutation could be associated with many cases of intellectual disability and movement disorders of unknown cause.
ObjectivesTo describe the characteristics of the patients identified in Spain with STXBP1 syndrome, and the implications for the diagnosis of these characteristics.
Patients and methodsThe details are presented on 17 individuals, aged between 2 years and 17 years, diagnosed in Spain with STXBP1 syndrome.
ConclusionsThere is a clear under-diagnosis of STXBP1 syndrome in Spain. Besides the inherent diversity of the disorder, with the increase in the number diagnoses the variability of the phenotype is even wider. The description of the alarm signs is necessary in order to identify those individuals with less prototypical manifestations of the disorder.
El síndrome STXBP1 es una alteración genética que tiene importantes implicaciones en el neurodesarrollo. El gen STXBP1 está relacionado con la regulación de los procesos que tienen lugar en la exocitosis, es decir, con los mecanismos que controlan la sinapsis. En concreto, este gen está implicado en el proceso de liberación de neurotransmisores por parte de las vesículas sinápticas. La sintaxin bindin protein, también conocida como Munc18-1, se une a la sintaxina formando un complejo que regula la liberación de neurotransmisores por parte de las vesículas sinápticas1-3.
En 2008 se describieron por primera vez mutaciones en el gen STXBP1 en pacientes con síndrome de Otahara o encefalopatía epiléptica infantil precoz4. Las descripciones de los pacientes incluían epilepsia de inicio muy temprano, dificultades motoras y retraso psicomotor. El gen STXBP1 aparece también vinculado a diversos síndromes epilépticos, como el síndrome de West, la epilepsia focal migratoria maligna del lactante, el espectro Dravet, la epilepsia mioclónica precoz grave, el espectro Rett con o sin epilepsia, el complejo autismo-epilepsia y la encefalopatía epiléptica discinética5. Según se han ido desarrollando estudios genéticos, el espectro de manifestaciones clínicas se ha ido ampliando progresivamente. Deprez et al.6, estudiando una muestra de niños con encefalopatía epiléptica infantil temprana que no mostraban síndrome de Otahara ni de West, encontraron mutaciones en STXBP1 en el 10% de los pacientes. Por otra parte, se han encontrado mutaciones en este gen en personas con trastornos del espectro del autismo (TEA) en una proporción significativamente mayor que en la población general7.
Aunque inicialmente la epilepsia parecía una característica definitoria del síndrome, progresivamente han aparecido descripciones de personas con la mutación que no han manifestado epilepsia en ningún momento. Hamdan et al.8 analizaron una muestra de pacientes con discapacidad intelectual no sindrómica y encontraron un caso de mutación en el STXBP1 en una persona de 21 años sin que hubiera experimentado epilepsia en ningún momento de su vida. Posteriormente, se han reportado más casos de pacientes con alteraciones del movimiento y discapacidad intelectual que presentaban mutaciones en el STXBP1 sin epilepsia9, lo que ha ampliado aún más la caracterización del síndrome. Aunque inicialmente se consideró esta mutación como uno de los principales causantes de encefalopatía epiléptica precoz, estudios posteriores han puesto de manifiesto que la epilepsia es una manifestación más de un amplio conjunto de síntomas que se expresan de manera muy variable. Se trata, por tanto, de un trastorno del neurodesarrollo y no de una encefalopatía epiléptica10-12.
Resulta necesario además realizar el diagnóstico diferencial con otras condiciones que generan procesos epilépticos de inicio precoz, como epilepsias dependientes de piridoxina, deficiencia en piridoxal 5’ fosfato oxidasa (PNPO), deficiencia cerebral de folato o deficiencias del transportador de glucosa tipo 1 (GLUT1)13.
Las descripciones clínicas de las personas con STXBP1 indican que, cuando aparece, la epilepsia se manifiesta de forma muy temprana. La mediana de la edad de inicio de las crisis se sitúa en las 6semanas de vida, en un rango que va desde el primer día de vida hasta los 12 años10. El control de las crisis suele ser costoso al inicio, aunque la evolución a este respecto es muy variable. Los estudios coinciden en señalar una buena evolución en gran parte de las personas con esta mutación, aunque una pequeña parte mantiene las crisis epilépticas de forma frecuente4-6,12. Stamberger et al.10 informan de que, en una muestra de 147 pacientes, cerca de la mitad (43,8%) dejó de presentar crisis entre el mes de vida y los 4 años, con la mediana situada en los 8 meses. Sin embargo, el 28,7% de la muestra seguía teniendo crisis de forma frecuente (más de una vez a la semana a pesar del tratamiento). También se informa de una reaparición de la epilepsia tras un largo periodo de remisión en 6de los pacientes estudiados. Todos los pacientes tenían discapacidad intelectual, en general con grandes necesidades de apoyo. Solo en un caso las dificultades cognitivas se limitaban a problemas de aprendizaje.
Cerca de la mitad de las personas identificadas por Stamberger et al.10 eran capaces de dar algunos pasos de forma independiente. Esta capacidad la desarrollaron entre los 14 meses y los 6 años. Un 15% de los pacientes tenían alguna capacidad de comunicación verbal y aparecía TEA en el 17% de la muestra. También se describen frecuentes dificultades de control del movimiento, como hipotonía, ataxia, temblor intencional, espasticidad discinesia y distonía10.
Esta variabilidad en las manifestaciones fenotípicas de la alteración puede conducir a un infradiagnóstico de la misma11. El seguimiento realizado en Dinamarca de una cohorte de 10 años indica una prevalencia de 1/91.86210. Hasta la fecha no contamos con datos de prevalencia del síndrome STXBP1 en España; sin embargo, los datos de la literatura hacen pensar que esta alteración podría estar relacionada con un gran número de trastornos del neurodesarrollo vinculados a alteraciones del movimiento y manifestaciones epilépticas.
El objetivo de este estudio es describir las características de la población con STXBP1 identificada en nuestro país, así como sus necesidades diagnósticas. La descripción de las características del síndrome, así como sus manifestaciones tempranas, pueden ayudar a mejorar la identificación de nuevos casos y el establecimiento de un diagnóstico cada vez más precoz.
Pacientes y métodoAl inicio del estudio existían en España 20 personas diagnosticadas de síndrome STXBP1 que hubieran contactado con la Asociación STXBP1, bien por propia iniciativa de las familias o derivados por los servicios diagnósticos. Se envió a las familias la información sobre el estudio, así como el consentimiento informado necesario en el caso de querer participar. La información recogida en este artículo pertenece a un estudio más amplio que tiene la finalidad de describir las características de este colectivo en España, definir sus necesidades y plantear vías prioritarias de actuación para mejorar su calidad de vida y la de sus familias. De las 20 familias contactadas, 17 accedieron a participar (85%). Una vez recopilados los consentimientos informados, se solicitó a las familias información a través de 3cuestionarios on-line que fueron enviados en 3momentos distintos. Los cuestionarios eran cumplimentados de forma anónima por uno o ambos progenitores. En estos cuestionarios se incluía información sobre la persona con STXBP1, las características familiares, así como sobre su experiencia y valoración personal de distintos procesos y facetas de la vida de la persona con STXBP1 y de su familia.
ResultadosDistribución geográficaHan participado en alguna fase del estudio 4familias de Barcelona, 3de Madrid, 2de Valencia y una de Alicante, Toledo, Jaén, Valladolid, Zaragoza, Málaga, León y Ávila.
La tabla 1 muestra la estimación de casos tomando como referencia los datos de prevalencia de Stamberger et al.10, proyectados en la población española de menores de 19 años (según datos del INE del 2017) y el número de casos reales identificados por comunidad autónoma.
Estimación de casos en la población española por comunidad autónoma y número de casos reales identificados
| Comunidad Autónoma | Estimación en menores de 19 años | Casos reales identificados |
| Andalucía | 20 | 2 |
| Aragón | 3 | 1 |
| Asturias | 2 | 0 |
| Baleares | 3 | 0 |
| Canarias | 4 | 0 |
| Cantabria | 1 | 0 |
| Castilla y León | 4 | 4 |
| Castilla-La Mancha | 5 | 1 |
| Cataluña | 17 | 4 |
| Valencia | 11 | 3 |
| Extremadura | 2 | 0 |
| Galicia | 5 | 0 |
| Madrid | 15 | 3 |
| Murcia | 4 | 0 |
| Navarra | 1 | 0 |
| País Vasco | 4 | 0 |
| La Rioja | 1 | 0 |
| Ceuta | 0 | 0 |
| Melilla | 0 | 0 |
| Total | 101 | 17 |
Según el INE, se producen en España en torno a 400.000 nacimientos al año, lo que supondría alrededor de 4 casos nuevos anualmente en nuestro país.
Características de las personas con STXBP1Sexo y edadDe los 17 participantes, 7 son mujeres (41%) y 10 son hombres (59%). El rango de edad comprende desde los 2 hasta los 17 años. La media de edad es de 8,3 años, con una desviación típica de 4,7 y una mediana de 7.
EpilepsiaUna de las principales características en las personas con STXBP1, y que con frecuencia es el elemento que inicialmente indica la existencia de una alteración, es la aparición de epilepsia. Como se observa en la figura 1, las personas de la muestra siguen una distribución similar a la recogida en la literatura científica al respecto.
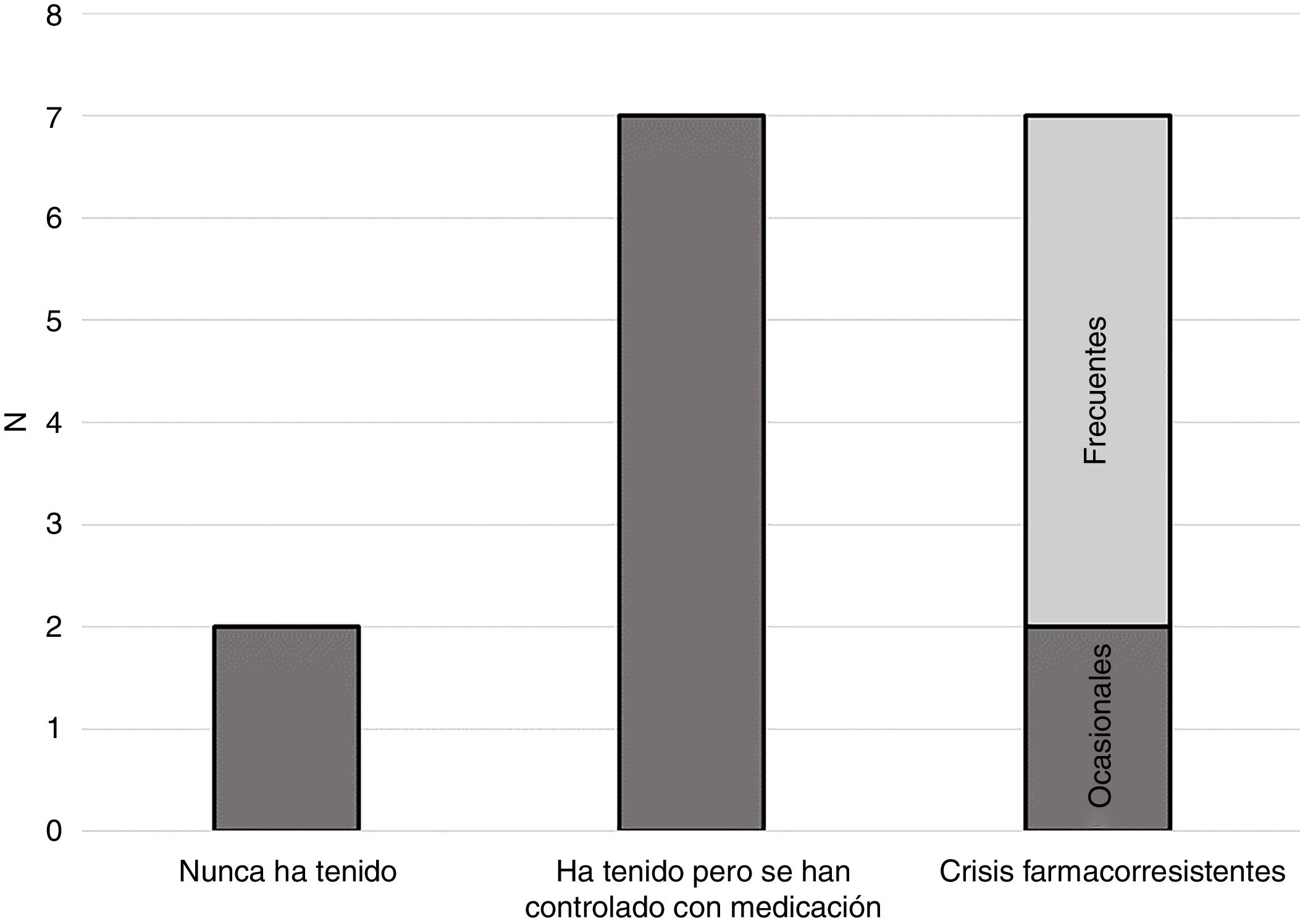
La mayoría de las personas con STXBP1 han experimentado crisis en algún momento, pero se han controlado con medicación (46%). Un 13% tiene crisis de forma ocasional (con una frecuencia menor que semanal) y un 33% tiene crisis frecuentes (con una frecuencia semanal o mayor). Una de las personas de la muestra nunca ha tenido crisis epilépticas. Otra tampoco ha tenido nunca crisis, pero sí una actividad electroencefalográfica anormal, concretamente se ha informado a la familia de la existencia de «picos epilépticos».
La epilepsia se manifiesta generalmente de forma muy temprana. La edad de inicio varía en un rango que comprende desde el primer día de vida hasta los 2años y 9meses. La mediana de la edad de inicio se sitúa en los 7 días. En cuanto a los tipos de epilepsia, se observa en nuestra muestra el amplio espectro de tipos de epilepsia que se menciona en la literatura previa. En la tabla 2 aparece el porcentaje de personas que han presentado cada uno de los tipos de epilepsia. Las manifestaciones más frecuentes son los espasmos epilépticos y las crisis generalizadas tónico-clónicas.
Tipos de epilepsia manifestados
| Tipos de epilepsia | % |
| Espasmos epilépticos | 71,4 |
| Crisis generalizadas: convulsiones tónico-clónicas | 71,4 |
| Crisis focales sin afectación del estado de consciencia | 50 |
| Crisis generalizadas: crisis de ausencia | 42,8 |
| Crisis generalizadas mioclónicas | 42,8 |
| Crisis focal (o parcial) con generalización secundaria | 28,5 |
| Crisis focales con afectación del estado de consciencia | 28,5 |
| Crisis generalizadas atónicas | 14,2 |
En cuanto al tratamiento farmacológico empleado, en todos los casos ha sido necesario probar más de un medicamento para el control de las crisis. Tres de las personas además siguen una dieta cetógena.
La mutación del gen STXBP1 aparece a veces ligada a otros síndromes que implican epilepsia; en nuestra muestra, una persona tiene un diagnóstico de síndrome de West, otra ha recibido un diagnóstico de síndrome de Otahara, 2personas más han recibido ambos (West y Otahara) y, por último, una persona ha recibido ambos diagnósticos y además el diagnóstico de síndrome de Dravet.
Grado de discapacidad reconocidoTodas las personas que han participado en el estudio tienen algún grado de discapacidad reconocido, desde un 40% hasta un grado de discapacidad reconocido superior al 90%, reflejando necesidades de apoyo de distinta intensidad.
Desarrollo motorEn cuanto al desarrollo motor, existe también una alta variabilidad en cuanto a las posibilidades de autonomía de las personas con STXBP1. Contamos con datos de 15 familias sobre este aspecto. En cuanto al desarrollo motor, el 60% de las personas tiene alguna capacidad de movimiento autónomo, ya sea en contextos limitados, con ayuda o para pequeños desplazamientos en el entorno próximo. El 40% no tiene posibilidades aún de desplazarse de forma autónoma. La figura 2 resume las capacidades de desplazamiento autónomo de las personas con STXBP1 participantes en el estudio.
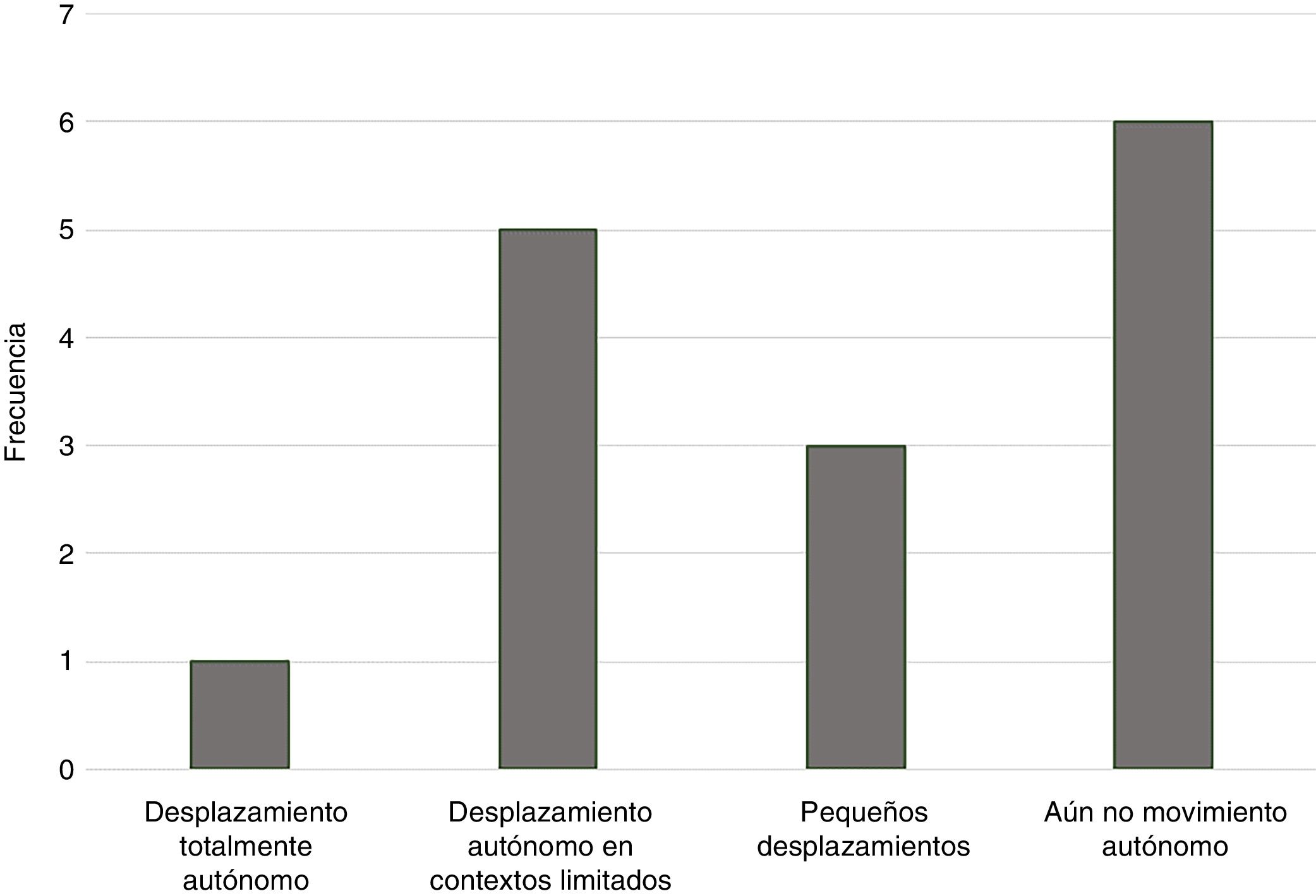
Esta capacidad de desplazamiento es totalmente autónoma en el 6% de los casos. Un 33% puede desplazarse de forma autónoma en contextos limitados, como, por ejemplo, la casa o el centro escolar, o con ayuda de alguien. Un 20% puede desplazarse de forma independiente pequeñas distancias, por ejemplo, gateando para alcanzar un juguete. Finalmente, un 40% de las personas no cuenta con ninguna forma de desplazamiento autónomo.
Otras características motorasAdemás del retraso psicomotor, la mayoría de las personas con STXBP1 cuenta con otras características motoras que dificultan la coordinación y el ajuste del movimiento voluntario. La figura 3 muestra la frecuencia de personas con STXBP1 que presentan rigidez, discinesia, ataxia e hipotonía. La suma de las frecuencias es superior a 15 porque algunas personas muestran más de una característica.
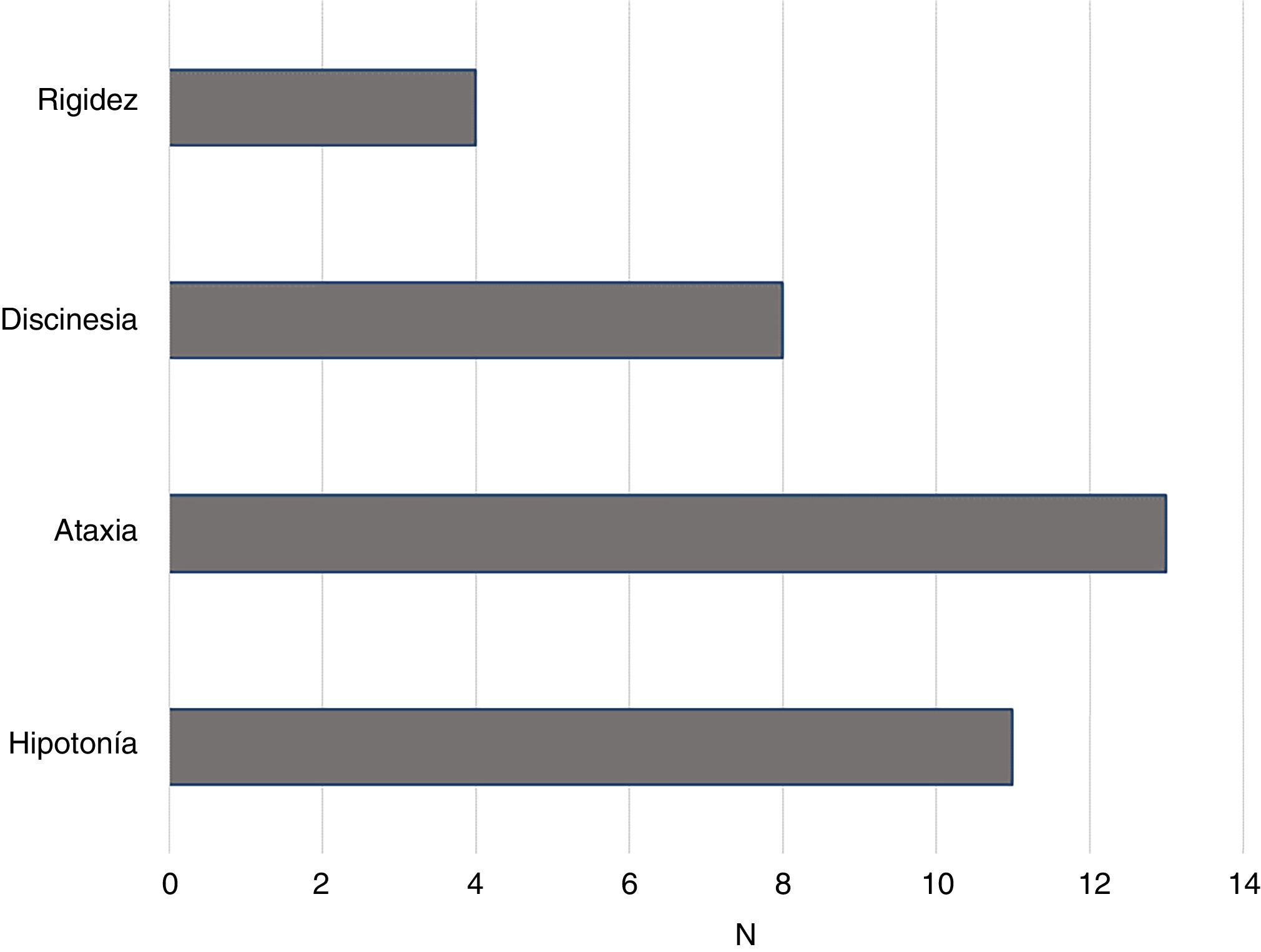
Como se observa en la figura 3, las características más frecuentes son la ataxia y la hipotonía. También aparece con frecuencia discinesia y en menor medida, rigidez.
Desarrollo comunicativo y socialA nivel comunicativo, la mayoría de los participantes (67%) se encuentra en una fase preintencional. Muestran todavía poca iniciativa por comunicar y es el entorno el que debe interpretar sus intenciones y deseos. Uno de los participantes tiene además un diagnóstico de TEA. El resto se distribuye entre personas que tienen intención por comunicar y emplean vocalizaciones, gestos o la mirada con fines comunicativos (13%), personas que utilizan palabras aisladas (13%) y personas que son capaces de construir frases para comunicarse (7%).
En cuanto al uso de sistemas aumentativos de comunicación, el 46% usa algún sistema aumentativo de comunicación. De estas personas, el 22% emplea signos, el 45% pictogramas y el 33% comunicadores. De las 11 personas que no cuentan todavía con lenguaje oral o comunicación vocal y han desarrollado por el momento una intención por comunicar limitada, 7 de ellas (63%) no cuentan con ningún sistema aumentativo de comunicación.
En lo relativo a las relaciones con los demás, la mayoría de las personas participantes en el estudio reconocen y reaccionan de manera diferencial a personas conocidas. Disfrutan con juegos interactivos sencillos y piden que continúen utilizando distintos medios. La mitad de los participantes muestra interés por los iguales y un 46% hace intentos por iniciar interacciones con iguales.
En lo que respecta a las relaciones sociales, cabe destacar la existencia de un diagnóstico de TEA y la vinculación con el TEA apuntada en la introducción. Las dificultades motoras y cognitivas pueden en ocasiones enmascarar dificultades específicas de relación social, por lo que es necesario indagar en este aspecto con mayor profundidad.
El proceso diagnósticoLas 15 familias que han completado esta fase del estudio conocen el diagnóstico hace menos de 5años. Siete de las familias conocían el diagnóstico desde hacía 6 meses o menos y las 11 restantes lo obtuvieron hace entre uno y 5años.
Los síntomas iniciales se han manifestado de forma temprana en todos los casos. La edad media de inicio de los primeros síntomas es de 3 meses, en un rango que oscila entre los primeros días de vida y los 13 meses de edad.
En la mayoría de los casos (10 de 15), los síntomas iniciales han tenido que ver con crisis epilépticas, espasmos o movimientos extraños que han hecho sospechar que algo no iba bien. Sin embargo, a pesar de la caracterización del síndrome como una encefalopatía epiléptica, en algunos casos los síntomas iniciales no han tenido que ver con episodios convulsivos, crisis o espasmos: a veces son dificultades de tipo motor, falta de progresión psicomotriz, signos neurológicos o llanto incontrolable. Lejos de ser algo anecdótico, en nuestra muestra el 33% de las familias informan de primeros síntomas de este tipo. En la tabla 3 se resumen los primeros signos de alarma detectados por las familias.
Primeros síntomas de las personas con STXBP1
| Primeros síntomas | N.° |
| Crisis epilépticas | 8 |
| Espasticidad y espasmos | 1 |
| Movimientos raros que no cesaban | 1 |
| Hipotonía | 2 |
| Falta de progresión motora (no se incorporaba, no se sentaba, no gateaba ni andaba) | 2 |
| Llanto incontrolable | 1 |
| Hiperexcitabilidad de reflejos tras el nacimiento | 1 |
El diagnóstico definitivo ha sido proporcionado a las familias por el neuropediatra en el 57% de los casos. Un 14% de las familias ha recibido el diagnóstico de un neurólogo y el 29% restante de un genetista. Los métodos empleados para la determinación del diagnóstico aparecen en la tabla 4.
A pesar de que, como decíamos, los síntomas iniciales se han manifestado de forma temprana en todos los casos, la obtención de un diagnóstico definitivo se ha dilatado considerablemente en el tiempo. La demora diagnóstica varía desde los 7 meses a los 14 años de espera.
Cuando consideramos los síntomas iniciales que llevaron a sospechar la existencia de algún problema en el desarrollo, observamos que la demora diagnóstica es mucho mayor en aquellos casos en los que los síntomas no incluyen crisis epilépticas, espasmos o movimientos extraños. La figura 4 muestra la demora diagnóstica media en función de si los síntomas iniciales incluyen crisis epilépticas o no.
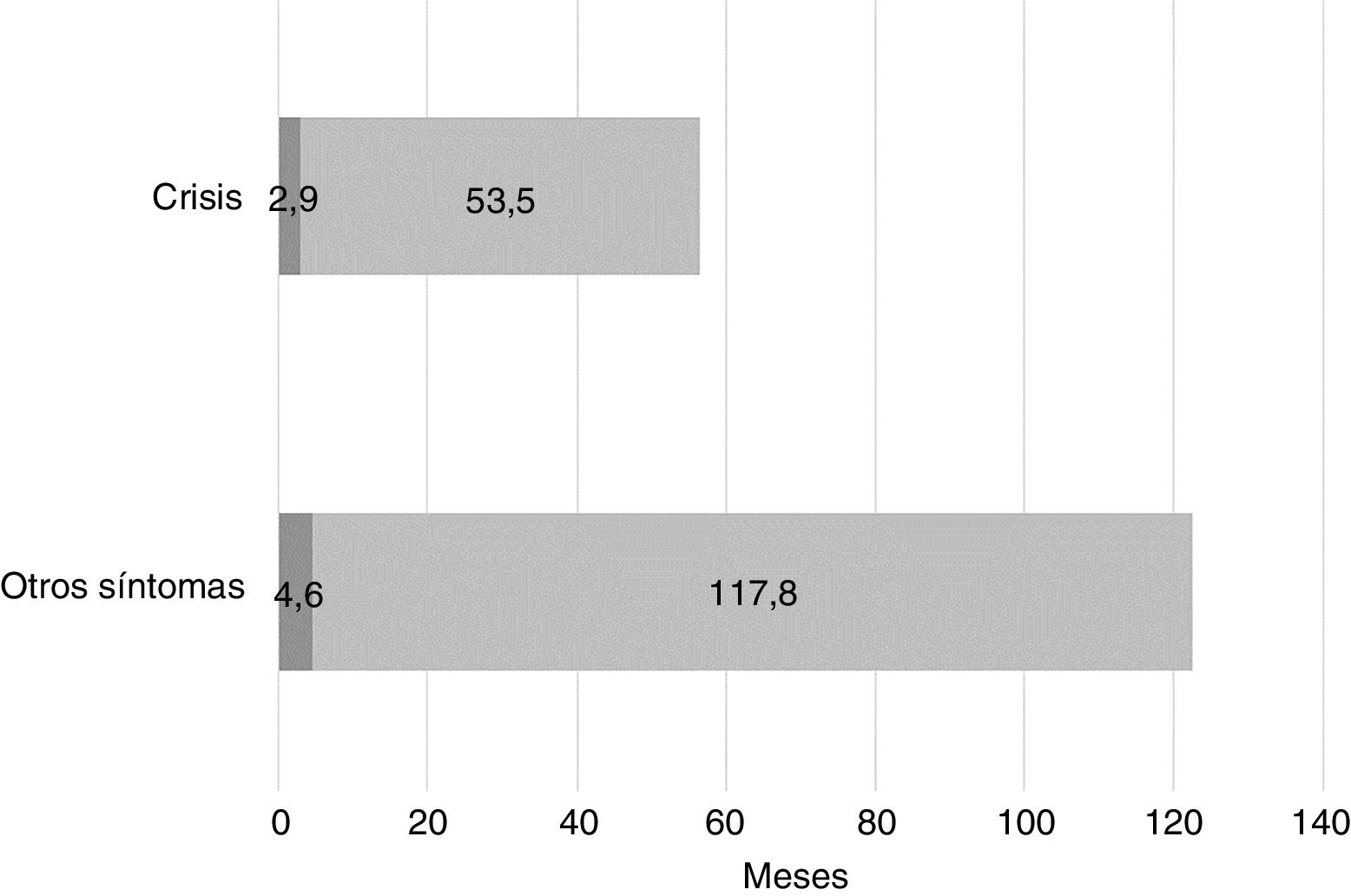
La valoración subjetiva de las familias sobre el proceso diagnóstico es acorde con los datos objetivos, indicando 2necesidades claras: 1) mayor rapidez en la obtención de un diagnóstico, de manera que se eviten tratamientos, pruebas y exploraciones innecesarias y se oriente cuanto antes el tratamiento, y 2) mayor información sobre la enfermedad, que facilite una mejor comprensión de los que está pasando y permita el establecimiento de redes de apoyo.
DiscusiónEl síndrome STXBP1 se manifiesta de forma muy temprana y tiene importantes implicaciones para el neurodesarrollo. El diagnóstico temprano es fundamental a la hora de establecer los apoyos necesarios, evitar pruebas innecesarias y orientar a las familias en la gestión de los recursos necesarios. Para favorecer la difusión de la información y la detección lo más temprana posible, se ha recopilado la información de 17 familias diagnosticadas en España sobre las características de las personas con STXBP1 y el proceso diagnóstico experimentado. La metodología empleada tiene sus limitaciones, ya que contamos con datos de 17 familias y la información ha sido recabada a través de las familias sin ser cotejada con informes médicos. Sin embargo, teniendo en cuenta la baja incidencia del síndrome y la relativa novedad de los diagnósticos, creemos que la información recopilada tiene un valor importante como punto de partida.
A continuación, se resumen las necesidades detectadas respecto del proceso diagnóstico.
En primer lugar, el número de casos de personas con STXBP1 identificados hasta ahora en España está muy por debajo del que sería esperable considerando los datos de prevalencia existentes. Teniendo en cuenta la distribución de la población en España y las provincias de residencia de las personas diagnosticadas, resulta evidente que los datos de detección están vinculados a la posibilidad de acceso a las pruebas o servicios diagnósticos necesarios, y no a la incidencia del síndrome. La desigual distribución geográfica de las personas diagnosticadas hace pensar en un importante infradiagnóstico en aquellas regiones que no cuentan con servicios especializados o con capacidad para realizar los análisis genéticos necesarios. Por otra parte, cada vez se conocen más características del fenotipo asociado a alteraciones en el STXBP1, como, por ejemplo, la existencia de casos con la mutación que no han presentado nunca alteraciones de tipo epiléptico9. Es, por lo tanto, esperable que con el aumento de la información sobre el síndrome, así como con una mayor facilidad de acceso a las pruebas diagnósticas, se incremente el número de casos identificados en nuestro país de forma muy notable. A pesar de que actualmente el síndrome STXBP1 es una enfermedad considerada ultrarrara, los datos indican que esta mutación podría estar detrás de múltiples casos de discapacidad intelectual, trastornos del movimiento y encefalopatías epilépticas de causa desconocida hasta el momento.
Actualmente, la demora hasta obtener el diagnóstico es muy elevada. Dicho diagnóstico se retrasa especialmente en los casos en los que no hay crisis epilépticas, espasmos o movimientos extraños como indicadores tempranos de una alteración. La falta de progresión psicomotriz, así como alteraciones de tipo motor, podrían suponer un punto de partida sobre el que investigar manifestaciones tempranas o signos de alarma que orienten hacia el análisis genético necesario.
La mayoría de las personas con STXBP1ha presentado o presenta epilepsia en algún momento de su vida. El avance en la investigación para el control de la epilepsia es fundamental, por un lado, para conseguir el control de las crisis en los casos en los que es difícil y, por otro lado, para conseguir reducir al mínimo posible la ingesta de medicación. Teniendo en cuenta que en algunos casos se da una reaparición de la epilepsia después de un largo periodo de remisión, resulta fundamental investigar qué indicadores biológicos y conductuales pueden proporcionarnos información sobre el patrón de evolución de la epilepsia. De este modo, podremos evitar la medicación innecesaria o, al contrario, mantener el tratamiento cuando haya riesgo de reaparición de las crisis.
Las características de las personas con STXBP1 de la muestra ponen de manifiesto la amplia variabilidad en cuanto a necesidades de apoyo en distintos ámbitos. Resulta fundamental la investigación en lo relativo al desarrollo comunicativo y lingüístico. Aunque en las investigaciones previas se informa de un 17% de personas con STXBP1 y además TEA, en nuestra muestra solo una persona ha obtenido este diagnóstico. Sin embargo, la mayoría de las personas de la muestra tienen poca iniciativa por comunicar y es el entorno el que debe interpretar sus intenciones y deseos. Aunque este aspecto requiere de una investigación en profundidad, no está claro que esta falta de iniciativa se deba únicamente a dificultades de tipo cognitivo o motor. Más bien al contrario, estas dificultades pueden enmascarar dificultades específicas de tipo comunicativo que sería necesario explorar. Es, por lo tanto, fundamental, investigar a este respecto y desarrollar programas de intervención y aplicaciones tecnológicas que garanticen el derecho a la comunicación de estas personas.
También es escaso el conocimiento existente sobre la eficacia de las distintas intervenciones centradas en favorecer el desarrollo motor en las personas con STXBP1.
La mejora en la detección y el diagnóstico, así como la investigación sobre tratamiento genético, médico y conductual, resultan fundamentales para mejorar la calidad de vida de estas personas y de sus familias.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Gracias a las familias de la Asociación STXBP1, por su inestimable colaboración en este estudio. Información y contacto con la asociación puede encontrarse en: https://stxbp1.es/
