



El síndrome de Guillain-Barré (SGB) es una polirradiculoneuropatía inflamatoria aguda, de origen autoinmunitario, desencadenada por una infección viral o bacteriana que produce una parálisis flácida y arrefléxica con afectación simétrica de inicio distal y avance proximal. Afecta a pacientes de todas las edades, desde la época de lactante hasta la vejez, siendo un trastorno relativamente frecuente en la edad pediátrica, representando la causa más común de parálisis flácida aguda en niños1.
Presentamos el caso de un niño asiático de 9 años de edad remitido por cuadro consistente en diplopía, náuseas y vómitos de 3 días de evolución. El día del ingreso asociaba debilidad muscular en extremidades superiores. Refería catarro de vías altas 10 días antes del inicio de la sintomatología. Antecedentes personales y familiares, sin interés.
En la exploración física presenta regular estado general, halitosis y dolor a la palpación del seno maxilar derecho. Neurológicamente destaca una ligera pérdida de fuerza en miembros superiores (iv/v), reflejos osteotendinosos vivos de forma generalizada con respuesta plantar flexora y discreta lateralización hacia la izquierda de la marcha.
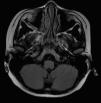
La resonancia magnética craneal mostraba ocupación de celdillas etmoidales con engrosamiento mucoso maxilar bilateral y esfenoidal de etiología inflamatoria (fig. 1). Se realizaron potenciales evocados visuales y examen oftalmológico que resultaron normales, así como el hemograma y la bioquímica general.
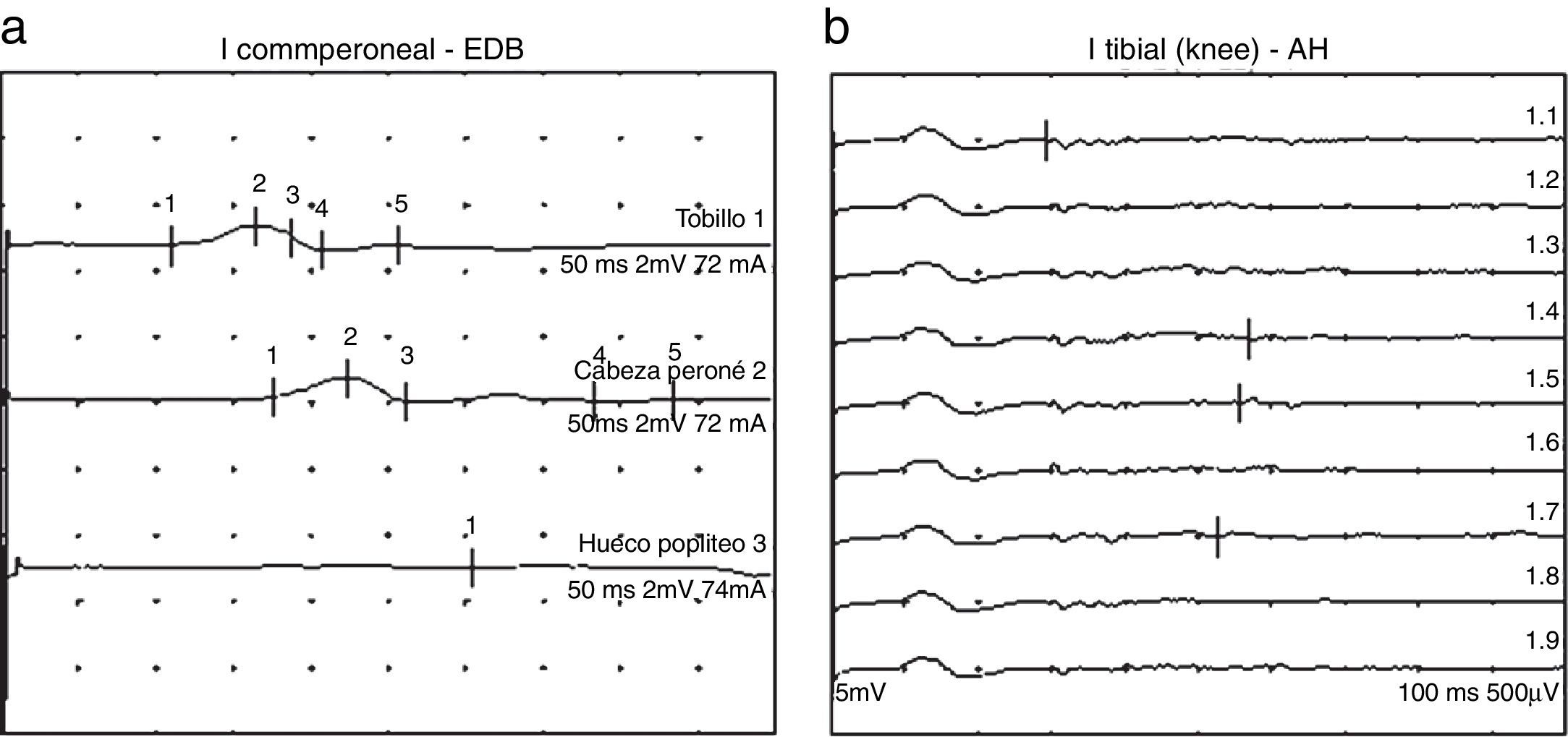
Se inició tratamiento antibiótico y tópico nasal. Durante los primeros 5 días asocia progresivamente estreñimiento, fotofobia y cefalea y, en las últimas 48 h, parálisis descendente con aparición de parálisis facial izquierda, ptosis palpebral bilateral, midriasis y disfagia. Ante la sospecha clínica de botulismo, y con el antecedente de ingesta de polvo seco de marisco, se administró antitoxina botulínica sin mejoría de la sintomatología. Se realizó entonces punción lumbar, detectándose hiperproteinorraquia con disociación albumino-citológica y electromiograma compatible con SGB (fig. 2). La serología para Campylobacter jejuni (C. jejuni), citomegalovirus, virus de Epstein-Barr, virus de la varicela zóster, Mycoplasma, Haemophilus influenzae, Borrelia budgorferi fue negativa.
Se inició terapia con inmunoglobulinas por vía intravenosa según protocolo (0,4 g/kg/día una vez al día durante 5 días), con mejoría clínica progresiva, recuperando la fuerza y la movilidad de las extremidades y desapareciendo la parálisis facial. La exploración física a los 15 días del alta hospitalaria es normal.
En la actualidad, se consideran 4 subtipos de SGB, determinados principalmente por sus características electrodiagnósticas y anatomopatológicas: polirradiculopatía aguda inflamatoria desmielinizante que es la más frecuente, neuropatía axonal sensitivo-motora aguda, neuropatía axonal motora aguda y síndrome de Miller-Fisher (SMF)2, caracterizado por oftalmoplejía, ataxia y arreflexia y relacionado con cepas de C. Jejuni que inducen la formación de anticuerpos antigangliósido GQ1b.
Los criterios diagnósticos necesarios para el SGB (adaptados de Asbury y Cornblath) son: a) pilar clínico; b) estudio del líquido cefalorraquídeo, y c) estudio electrofisiológico. Clínicamente, son características la debilidad progresiva en 2 o más miembros por neuropatía, la arreflexia, la evolución de la enfermedad en menos de 4 semanas, la disfunción autonómica y la exclusión de otra causa, como por ejemplo vasculitis, toxinas, botulismo, miastenia gravis, difteria, porfiria, meningoencefalitis, compresión medular, mielitis transversa, miositis aguda, etc.3. Hay también criterios diagnósticos clínicos de apoyo, como la debilidad relativamente simétrica, la afección sensitiva de grado leve, la ausencia de fiebre, el perfil típico en el líquido cefalorraquídeo (disociación albumino-citológica) y signos electrofisiológicos de desmielinización4.
El SGB ha sido ampliamente recogido en la literatura especializada. En casos leves, se ha descrito como debilidad simétrica de las extremidades inferiores, con reflejos osteotendinosos disminuidos o ausentes hasta llegar a causar en los casos más graves compromiso respiratorio e incluso la muerte. El diagnóstico en niños suele retrasarse, en parte por la presentación clínica inespecífica, el examen neurológico desafiante y los posibles diagnósticos alternativos5–7.
Además, hasta un 11% de los casos pediátricos pueden presentarse como variantes atípicas del SGB por no cumplir los criterios de Asbury8.
La forma de presentación de nuestro caso no es la habitual. Tras un periodo inicial con diplopía, aparece un cuadro caracterizado por fotofobia, estreñimiento, cefalea y, lo más llamativo, una parálisis descendente con parálisis facial izquierda y disfagia. A pesar de que en nuestro paciente se encontró como comienzo una afectación de nervios craneales, frecuente en el SMF (hasta el 50% de los casos)3, no se consideró como tal, bien por la negatividad de la serología para C. jejuni, pero sobre todo por la ausencia de ataxia y arreflexia.
Sabiendo que existen variantes atípicas del SGB, es fundamental elevar el umbral de sospecha y tener presente que «es siempre más frecuente un síntoma raro de una enfermedad frecuente que un síntoma frecuente de una enfermedad rara».
