


La hiperplasia suprarrenal congénita por déficit de 21-hidroxilasa (21OHD) es una enfermedad monogénica de herencia autosómica recesiva. El análisis molecular del gen CYP21A2 (OMIM +201910) permite confirmar el diagnóstico, identificar portadores y realizar diagnóstico prenatal1. Ante un caso índice resulta fundamental proporcionar un consejo genético correcto y adecuado a cada familia.
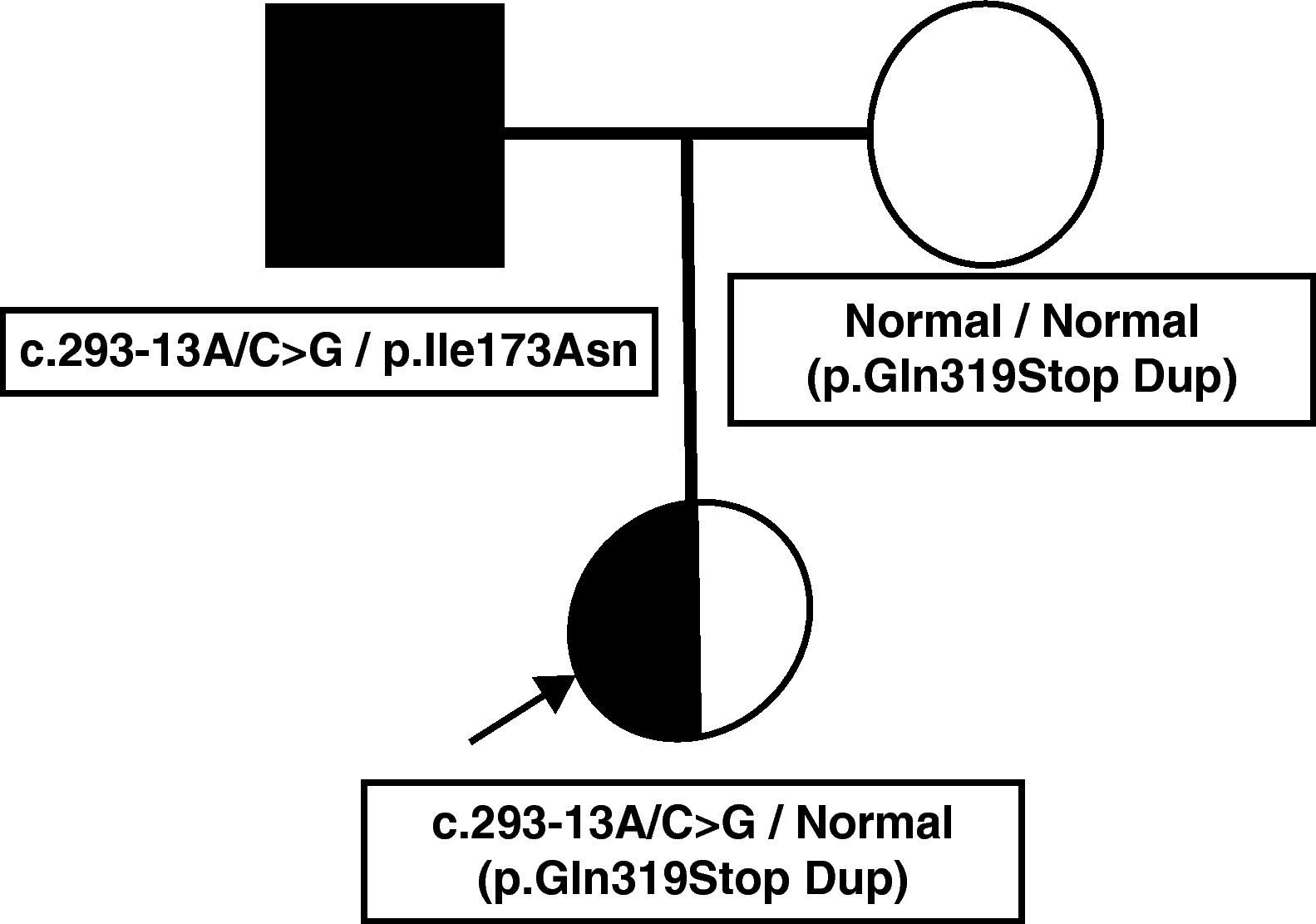
Presentamos el caso clínico de una niña de 6 días de vida, asintomática, referida por ser hija de varón diagnosticado de 21OHD. La niña era producto de una gestación planificada. El padre había sido diagnosticado de 21OHD a los 4 años tras presentar pubarquia y aceleración de la velocidad de crecimiento y en la actualidad seguía tratamiento con dexametasona y fludrocortisona. No se le había realizado estudio genético. La madre aportaba una determinación de 17-hidroxiprogesterona (17OHP) tras estímulo con corticotropina (ACTH) normal, realizada previamente al embarazo. El embarazo y el parto fueron normales, con una somatometría normal al nacimiento. La exploración física fue normal. La determinación de 17OHP en la muestra de sangre capilar procedente del cribado neonatal fue 12 nmol/l (normal<30 nmol/l), pero dados los antecedentes familiares, se realizó una determinación de 17OHP sérica que se encontró levemente por encima del límite normal y estudio genético mediante el análisis directo del gen CYP21A2 a través del estudio de deleciones y conversiones por técnica de Southern y el cribado de las mutaciones puntuales recurrentes mediante PCR e hibridación específica de alelo. Dicho estudio mostró las mutaciones graves c.293-13A/C>G (655G) y p.Gln319Stop (Gln318Stop). Sin embargo, al realizar el estudio de la dosis génica según la técnica previamente descrita por Ezquieta et al2 se detectó que la paciente presentaba una duplicación del gen funcional CYP21A2. La segregación de alelos permitió documentar que el padre era heterocigoto compuesto para las mutaciones graves c.293-13A/C>G y p.Ile173Asn (Ile172Asn) y que la madre presentaba la mutación p.Gln319Stop en heterocigosis. El estudio del ADN de la madre permitió establecer que el alelo materno era el portador de duplicación del gen (p.Gln319Stop Dup) y, por tanto, se trataba de una variante normal no portadora de deficiencia. La niña fue diagnosticada como portadora de la mutación severa c.293-13A/C>G (fig. 1).
Como se deduce de lo expuesto, todo caso índice de una enfermedad monogénica debe recibir un consejo genético adecuado. El estudio ha de extenderse a su familia y, en el caso de desear descendencia, a la pareja. La alta tasa de portadores para 21OHD en la población general (1:60 para mutaciones graves) hace que un paciente con una forma clásica tenga una probabilidad de tener un descendiente con una forma grave de 1:120 si el genotipo de la pareja es desconocido1. La prueba diagnóstica más adecuada para la identificación de portadores es el análisis genético1,3, ya que los portadores pueden presentar valores de 17OHP tras estímulo con ACTH normales y por la imposibilidad para discernir si se trata de un portador de un alelo grave o leve a través de los niveles de 17OHP4.
Nuestro caso demuestra además la necesidad de realizar el estudio genético para la 21OHD con las técnicas más avanzadas para no facilitar consejos genéticos inadecuados. La existencia de un seudogén con un 98% de homología y su localización en una región muy pleomórfica predisponen a la aparición de recombinaciones asimétricas durante la meiosis, resultando en deleciones, duplicaciones y transferencia de mutaciones desde el seudogén5. Deben tenerse en consideración las variantes que, en estudios incompletos, pueden mostrarse como aparentes alelos graves o leves, especialmente si no son infrecuentes2,6, como cuando se produce la mutación en un gen CYP21A2 duplicado, lo que ocurrió en nuestra paciente.
La prevalencia de duplicaciones del gen CYP21A2 en la población general oscila entre el 1,67 y el 7%8 y tiene especial relevancia en el caso de la mutación p.Gln319Stop (se ha descrito que el 84% de los individuos con la mutación p.Gln319Stop presentan duplicación del gen9). En nuestro caso se empleó el método basado en PCR semi-cuantitativa, que permite la distinción entre ambas variantes alélicas (la mutación severa p.Gln319Stop y la variante con la duplicación), realizando una estimación semicuantitativa del número de copias del gen (método propuesto por Ezquieta et al2).
La principal conclusión a extraer del caso presentado es que el momento óptimo para la determinación del riesgo genético, clarificación del estado de portador y la discusión de la disponibilidad de diagnóstico y tratamiento prenatal es antes de la concepción, basado en el diagnóstico molecular de la 21OHD en un laboratorio de referencia. En nuestro caso, si la madre, en lugar de presentar una mutación asociada a una duplicación del gen, hubiera presentado únicamente la mutación, nuestra paciente hubiera presentado una forma clásica de 21-OHD, lo que implicaría una virilización de los genitales externos y la posible aparición de un síndrome pierde sal grave en el período neonatal. En ese caso nuestra paciente no habría podido beneficiarse del tratamiento prenatal con dexametasona, tratamiento experimental para evitar la virilización de los genitales, pero que debe iniciarse en el momento que se confirma la gestación (antes de la octava semana) para que sea eficaz y, por tanto, debe de plantearse en la consulta preconcepcional.
