


Sr. Editor:
El déficit del cofactor molibdeno es una enfermedad metabólica infrecuente, que se transmite de forma autosómica recesiva1. Es causa de síntomas neurológicos graves, fundamentalmente convulsiones neonatales refractarias, encefalopatía, microcefalia, hiperekplesia2, rasgos dismórficos, litiasis renal y luxación del cristalino3. Los estudios de imagen muestran atrofia cerebral o múltiples cavidades quísticas4 El pronóstico es malo, y los pacientes fallecen en los primeros meses. Ninguno de los tratamientos ensayados ha mostrado una eficacia clara (dieta exenta de aminoácidos sulfurados5, tetrahidrobiopeterina, D-penicilamina, cofactor molibdeno).
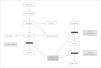
El molibdeno es un oligoelemento esencial para el funcionamiento de tres enzimas: sulfito oxidasa, xantina deshidrogenasa y aldehído oxidasa. Este déficit enzimático es el responsable de las manifestaciones más importantes. La neuropatogenia no ha sido aclarada. Por otra parte, el déficit de xantina deshidrogenasa afecta al metabolismo de las purinas y produce hipouricemia y aumento de xantina e hipoxantina en la orina (fig. 1). Estos hallazgos combinados deben sugerir el diagnóstico.
Nuestro paciente nació a término tras cesárea electiva con peso adecuado y Apgar 9/10. Tenía una hermana que falleció tras un cuadro de encefalopatía epiléptica precoz que en la tomografía computarizada presentaba atrofia córtico-subcortical, hipoplasia del vermis cerebeloso e imágenes quísticas en la sustancia blanca. La madre había sufrido tres abortos. No había consanguinidad ni otros antecedentes de interés.
Durante las primeras 24h presentó depresión neurológica, taquipnea, ausencia de reflejos arcaicos y dificultad para la alimentación, mioclonías palpebrales y de extremidades, y crisis de rigidez generalizada. Mostraba un occipucio prominente, micrognatia, un cuello corto con piel redundante, hipotonía axial y ausencia de tracción cefálica.
El electroencefalograma mostraba patrón de brote-supresión y la resonancia magnética (RM) cerebral revelaba una hipoplasia del vermis cerebeloso.
Los estudios realizados inicialmente, incluyendo el estudio del metabolismo intermediario, aminoácidos en sangre, orina y líquido cefalorraquídeo, biotinidasa, pterinas, CDG, carnitinas y acilcarnitinas, ácidos orgánicos en orina, test de sulfitos en orina, fueron normales. Destacaba la presencia de ácido úrico bajo (0,8mg/dl). El cariotipo también fue normal.
Posteriormente se realizó un nuevo test de sulfitos en orina, que fue positivo en esta ocasión, y además se detectó en el plasma un aumento de sulfocisteína de 54μmol/l (controles < 25), de xantina de 18μmol/l (controles 2,2 ± 1,3), de hipoxantina de 6μmol/l (controles 5,9 ± 4) y de taurina de 38μmol/l (controles 95 ± 54), una cistina indetectable (controles 18 ± 10μmol/l) y una homocisteína total de 0,7μmol/l (controles 6,3 ± 2,4), hallazgos compatibles con un déficit del cofactor molibdeno.
La actividad de la enzima sulfito oxidasa en fibroblastos era indetectable.
Mediante estudios genéticos se identificó la mutación c533-536 del GTCA en el gen MOCS2B en homocigosis.
El paciente evolucionó desfavorablemente, con hipotonía axial, tetraparesia, microcefalia, problemas de succión-deglución, litiasis renal y crisis. Una nueva RM mostró atrofia supra e infratentorial, del vermis cerebeloso, e hipoplasia del cuerpo calloso. Existían lesiones de encefalopatía multiquística (fig. 2). El paciente falleció a los 15 meses.
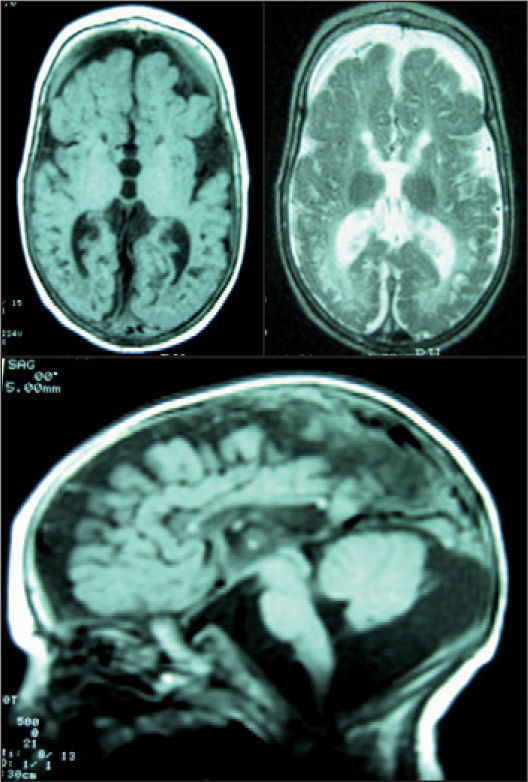
RM craneal. Se observa hipolasia casi completa del cuerpo calloso, marcada dilatación atrófica del sistema ventricular, atrofia cortical y central y un llamativo aumento del espacio extraaxial. Afectación completa de la mielina. Hipoplasia cerebelosa. Atrofia particular de los temporales con gran aumento de las cisternas subaracnoideas. No se identifican los principales ganglios de la base. Aspecto neurorradiológico de cerebro "en nuez".
El déficit del cofactor molibdeno es una enfermedad rara, de herencia autosómica recesiva, descrita por Duran et al en 19786 Los genes afectados responsables son: MOCS1, MOCS2 y GEPH7.
Se ha observado una variabilidad en la gravedad y la edad de comienzo, pero suele iniciarse en el período neonatal inmediato con dificultades para la alimentación y crisis convulsivas refractarias. Hay casos de comienzo más tardío, aunque con menor frecuencia que en el déficit aislado de sulfito oxidasa. Presentan un fenotipo peculiar, con la cara alargada, mejillas gruesas, hipertelorismo, hendiduras palpebrales largas, labios gruesos, filtro amplio y nariz pequeña.
Las alteraciones neurorradiológicas consisten en pérdida progresiva de la sustancia blanca, y pueden mostrar cavidades quísticas que recuerdan a la encefalopatía hipóxico-isquémica perinatal. Debe considerarse el diagnóstico en los casos de crisis rebeldes en el período neonatal, y clínica y radiología sugestivas de encefalopatía hipóxico-isquémica, sin antecedentes perinatales compatibles. Otras alteraciones descritas son atrofia cortical y cerebelosa, dilatación ventricular, calcificaciones focales, etc8.
Además de la hipouricemia marcada y el test de sulfitos en orina positivo, los pacientes presentan concentraciones descendidas o indetectables de homocisteína plasmática total9,10. Es un hallazgo típico del déficit aislado de sulfito oxidasa y del déficit del cofactor molibdeno, y una herramienta útil propuesta por algunos como la prueba inicial de cribado. En nuestro paciente se confirmó la hipohomocistinemia retrospectivamente.
La detección de sulfitos en la orina mediante tira reactiva es una forma rápida y sencilla, pero los sulfitos urinarios son muy inestables, por lo que pueden dar resultados negativos falsos. Los tiosulfatos y la S-sulfocisteína son más estables, pero su determinación no está al alcance de todos los laboratorios.
Cuando se sospecha esta enfermedad, se debe proceder a realizar estudios enzimáticos y/o genéticos con el fin de poder tratar mejor al paciente, y de realizar ulteriores diagnósticos prenatales.
Al doctor J. Reiss, del Institut fur Humangenetik, Universitats Kliniken, Gottinger, Alemania; doctor C. Dorche, Hopital Debrousse, Lyon, Francia; doctora. M. Rodes, Instituto de Bioquímica Clínica, Barcelona, España; doctora Ruidor, Hospital Vall d'Hebron, Barcelona, España; doctor A. Muñoz, Hospital 12 de Octubre, Madrid, España.
