



La lipodistrofia generalizada congénita o síndrome de Berardinelli-Seip es una entidad rara con herencia autosómica recesiva que se caracteriza por una ausencia congénita de tejido adiposo y secundariamente un defecto en la síntesis de leptina. Los sujetos afectos presentan un fenotipo clásico con rasgos acromegaloides, atrofia generalizada del tejido graso subcutáneo, apariencia musculada, acantosis nigricans, hepatomegalia y abdomen prominente. Desde el punto de vista metabólico, y como consecuencia del déficit de la leptina son características la hipertrigliceridemia marcada, que suele conllevar esteatosis hepática y la resistencia a la insulina. Presentamos el caso de dos sujetos de distintas familias afectos de lipodistrofia generalizada congénita que durante su evolución desarrollaron diabetes mellitus con mal control que fueron tratados con dosis elevadas de insulina y que presentaron precozmente complicaciones microvasculares. Se constató la existencia de mutación para el gen LMNA en uno de los sujetos.
Generalized congenital lipodystrophy or Berardinelli-Seip Syndrome is a rare autosomal recessive condition characterized by the absence of adipose tissue and eventually a defect in leptin synthesis. Affected subjects tend to show a classical phenotype with acromegaloid appearance, generalized atrophy of subcutaneous adipose tissue with muscular hypertrophy, acanthosis nigricans, hepatomegaly and prominent abdomen. From metabolic point of view and as a consequence of leptin absence, hypertriglyceridemia leading to hepatic steatosis and insulin resistance may appear. Two cases of unrelated subjects affected of generalized congenital lipodystrophy are presented. Both developed difficult-to-manage diabetes mellitus and were treated with high doses of insulin. In both cases early microvascular complications were present. A mutation for LMNA gene was found in one of the subjects.
Las lipodistrofias congénitas representan un grupo heterogéneo de enfermedades caracterizadas por alteraciones generalizadas o parciales del desarrollo y la distribución de la grasa corporal. La fisiopatología de la mayoría de lipodistrofias humanas continúa siendo desconocida; sin embargo, existe evidencia de que distintas alteraciones genéticas primarias son responsables de la alteración del desarrollo del tejido graso y la generación de las distintas alteraciones metabólicas. Atendiendo a la distribución corporal las lipodistrofias congénitas pueden clasificarse en generalizadas o parciales. La clasificación actual comporta distintos tipos de lipodistrofia congénita generalizada según el gen implicado: lipodistrofia congénita generalizada (LCG) tipo 1 (gen AGPAT2), LCG tipo 2 (gen BSCL2), LCG tipo 3 (gen CAV1) y LCG tipo 4 (gen PTRF). La lipodistrofia congénita familiar parcial tipo 2 tiene como gen candidato a LMNA, que codifica la proteína laminina A/C, si bien en algunos casos, mutaciones del gen PPAR-γ (peroxisome-proliferator-activated receptor γ) también han sido implicadas.
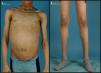
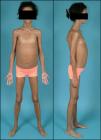
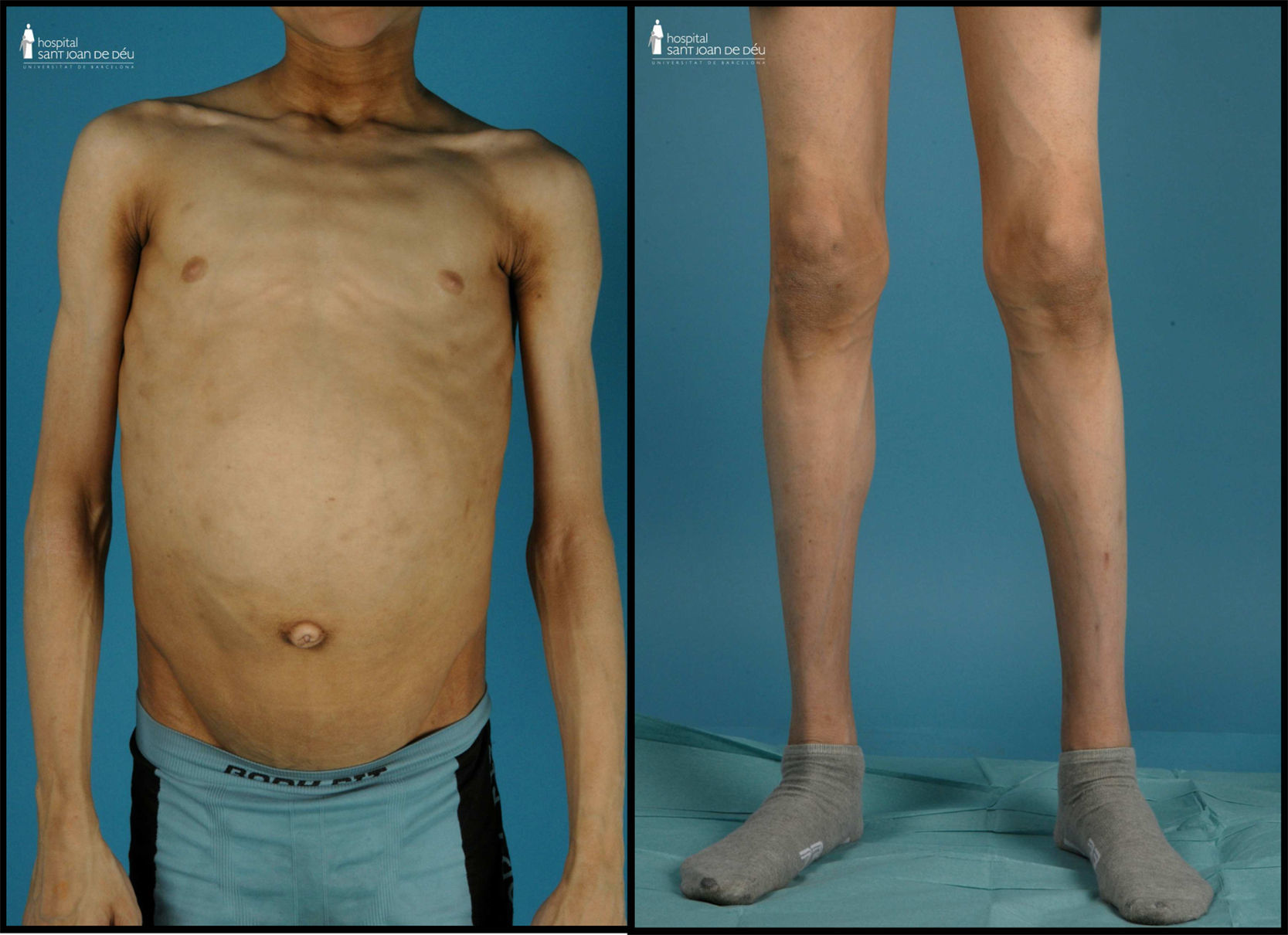
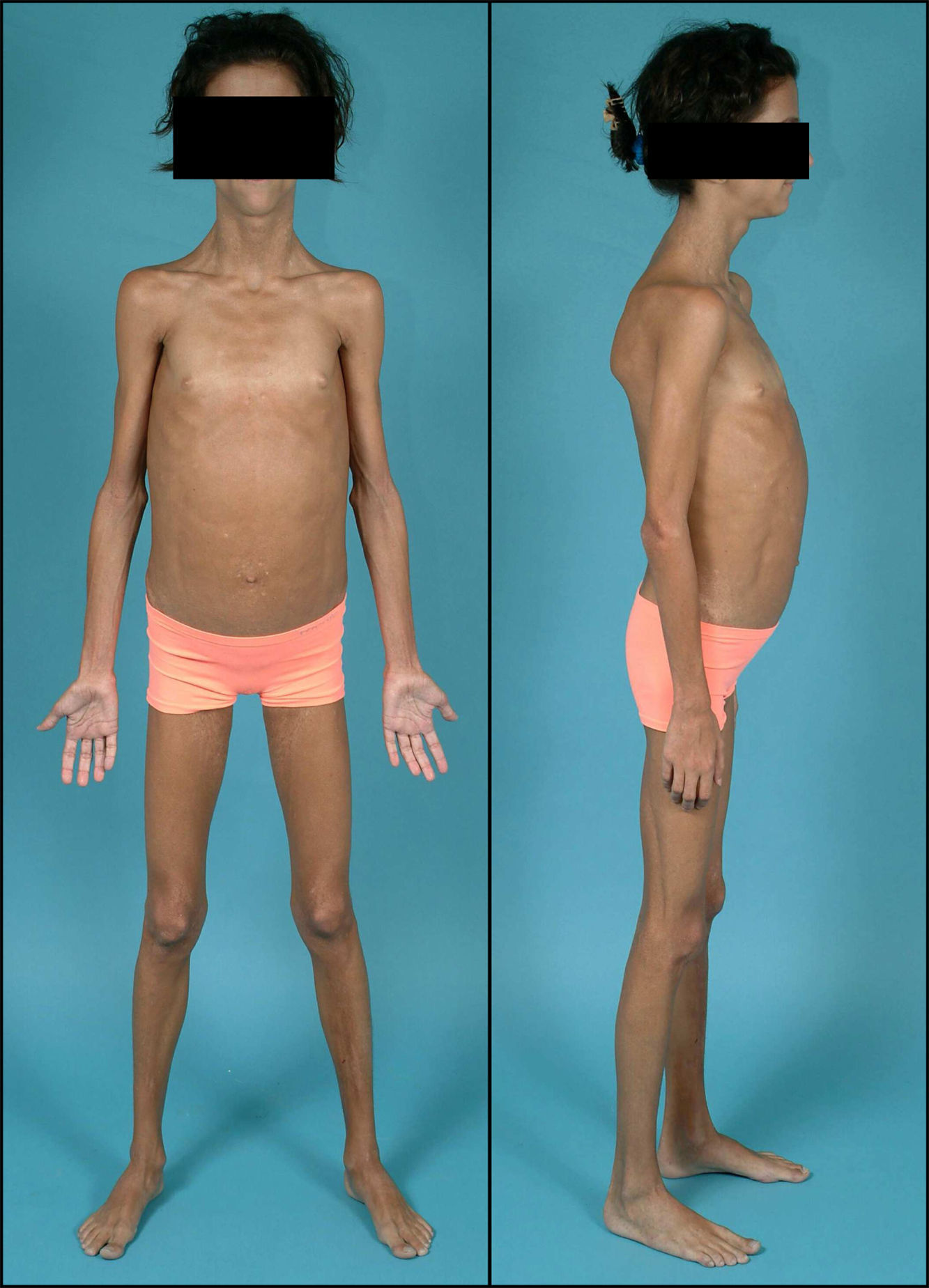
La LCG o síndrome de Berardinelli-Seip es una rara enfermedad de herencia autosómica recesiva que se caracteriza por una ausencia casi total de tejido adiposo desde el nacimiento1,2. Fue originalmente descrita por Berardinelli (1954) y Seip (1959) como un trastorno metabólico con lipoatrofia y alteraciones endocrinas. Como consecuencia de la ausencia de tejido adiposo hay un déficit de producción de leptina que condiciona una resistencia a la insulina muy marcada y un desarrollo precoz de diabetes mellitus, a menudo con presentación rápida de complicaciones microvasculares asociadas3. Los individuos afectos muestran un fenotipo característico con un marcado aspecto musculado, rasgos acromegaloides, acantosis nigricans, hepatomegalia y abdomen prominente. En algunos pacientes se ha descrito miocardiopatía hipertrófica y retardo mental leve4,5.
Casos clínicosCaso 1Paciente de sexo masculino de 13 años procedente de Arabia Saudí, hijo de padres consanguíneos de segundo grado que acude a urgencias por hiperglucemia >400mg/dl. El paciente no refería seguimiento médico en nuestro país desde su llegada hacía 9 meses. Durante este tiempo no había presentado ningún síntoma cardinal de diabetes mellitus. Refería como antecedentes de interés tiroiditis crónica autoinmune en tratamiento con preparado farmacológico de levotiroxina y triyodotironina, glomerulonefritis proliferativa-mesangial con semilunas y macroproteinuria en tratamiento con ramipril y cuadros de bronquitis de repetición por disfunción mucociliar.
Exploración física (fig. 1): peso: 50kg (-0,42 DE). Talla: 173cm (+1,09 DE); índice de masa corporal (IMC): 16,71kg/m2 (-1,19 DE). Tensión arterial: 102/53 mmHg. Lipodistrofia generalizada, con importante acantosis nigricans en región retrocervical y axilar así como xantoma duro en región inframandibular izquierda. Hepatomegalia blanda de 8cm. Genitales prepuberales. En la analítica sanguínea llevada a cabo destacaba: glucemia 706mg/dl; cetonemia 0,1 mmol/l; pH: 7,35; péptido C 1,21 mmol/l, anticuerpos anti-GAD 1225 UI/ml (+) y antiIA2 0,23 Ul/ml (-), creatinina 1,24mg/dl; colesterol total 244mg/dl; colesterol LDL 94mg/dl; triglicéridos 552mg/dl; hemoglobina glucada (HbA1C) 9,4%; insulina 18,5 mU/l; Se decidió iniciar pauta basal-bolus de insulina LysPro y glargina (1 U/kg/día) que se fue ajustando en función de los controles de glucemia capilar, precisando cantidades de insulina mayores a 2 U/kg/día. Así mismo se añadió al tratamiento metformina (1.700mg/día) y benzafibrato para control de la hipertrigliceridemia. Se efectuó examen de fondo de ojo que fue normal y analítica de orina que evidenció datos de hiperfiltración (aclaramiento de creatinina 137ml/min/1,73m2) y aumento del índice albúmina/creatinina (índice Alb/Cr 74mg/mmol).
En los meses posteriores ha persistido mal control metabólico con HbA1C >14% y requerimientos de insulina superiores a 4 U/kg/día que se han mantenido hasta el momento actual. Dado que el paciente cumplía criterios clínicos de LCG, se realizó determinación sérica de leptina que resultó ser anormalmente baja (<0,2mg/dl). El estudio genético no ha mostrado alteraciones en los genes BSCL2 ni AGAPT2. A la edad de 14 años y 7 meses presentó episodio de pancreatitis aguda coincidente con cifra de triglicéridos séricos elevada (5.836mg/dl) que se resolvió con éxito tras realización de plasmaféresis.
Caso 2Paciente mujer de 8 años y 11 meses remitida a consulta de Endocrinología para valoración de hiperglucemia asociada a dislipemia. Refería como antecedentes lesiones hiperpigmentadas en cuello, axilas, ingles y rodillas que presentaba desde el primer mes de vida y desde entonces en seguimiento por Dermatología que fueron clasificadas inicialmente como síndrome progeriforme. Un año antes de ser remitida a Endocrinología, se había iniciado seguimiento en Nutrición por hallazgo casual hipertrigliceridemia de 292mg/dl en tratamiento con benzafibrato, gemfibrozilo y ácidos omega 3. Igualmente se constató glucemia basal en ayunas alterada (113mg/dl) motivo por el cual se derivó a Endocrinología.
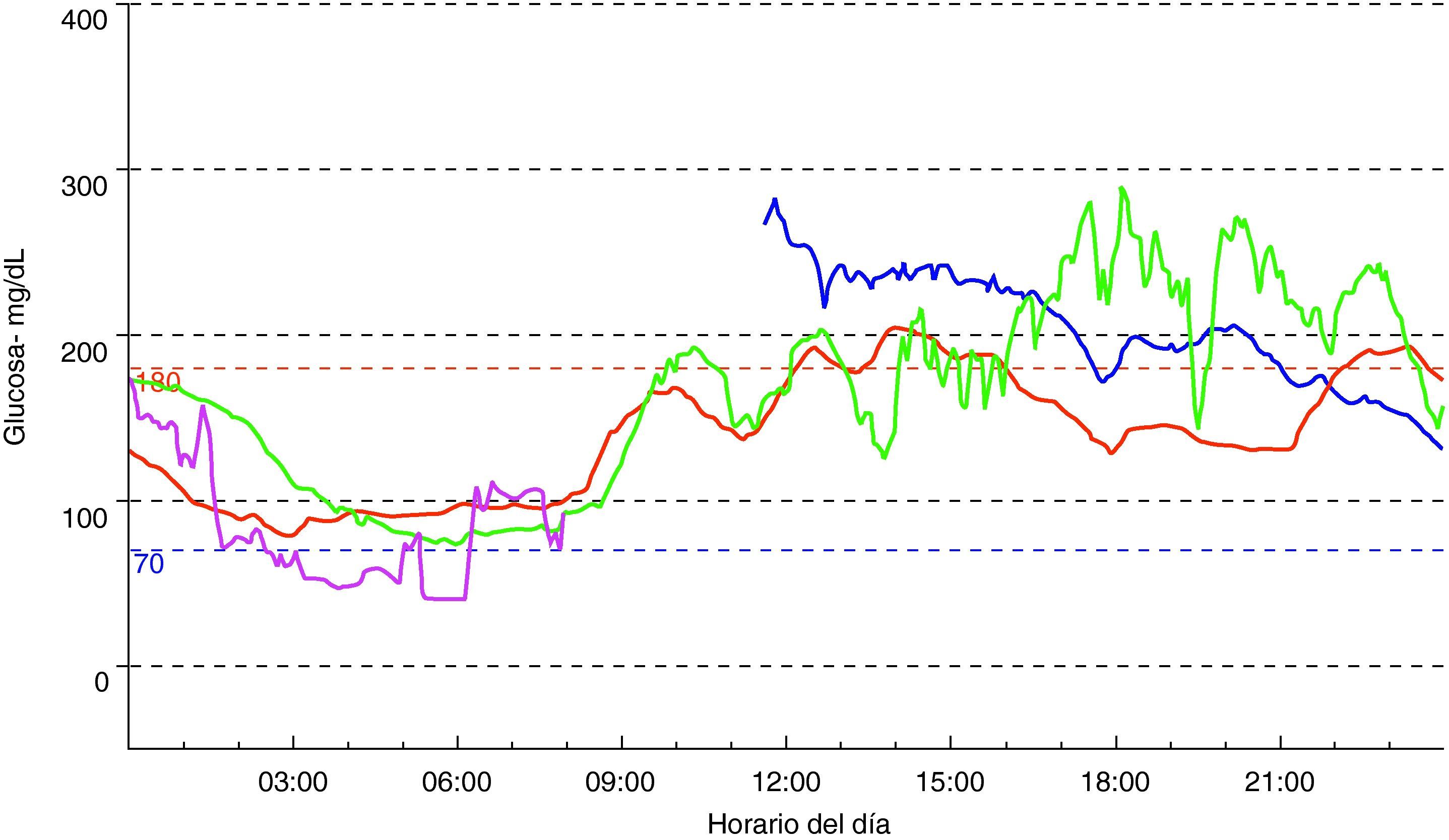
Exploración física (fig. 2): peso: 28kg (-0,55 DE); talla: 141,1cm (+1,7 DE); IMC: 14,08kg/m2 (-1,44 DE); tensión arterial: 105/59 mmHg. Aspecto distrófico por atrofia generalizada del tejido adiposo subcutáneo, hipertrofia muscular importante y hepatomegalia de 9cm. Exámenes complementarios: test de tolerancia oral a la glucosa: glucemia basal 112mg/dl; insulinemia basal 290 mU/l; péptido C basal 5,5 nmol/l; glucemia 120min 288mg/dl; insulinemia 120min 1069 mU/l; péptido C 120min 11,6 nmol/l; La determinación de anticuerpos anti-GAD y anti-IA2 resultó ser negativa con valores de péptido C >1,5 ng/ml. Se decidió iniciar tratamiento con acarbosa, presentando en ese momento buena respuesta. Al año de iniciar dicho tratamiento sufrió deterioro del control metabólico con glucemias superiores a 300mg/dl (fig. 3) y HbA1C de 8,7%. En ese momento se sustituyó acarbosa por insulina (glargina y LysPro). A los 11 años y coincidiendo con el inicio del desarrollo puberal presentó empeoramiento del control glucémico (HbA1C 10%), aumento de necesidades de insulina (1,3 U/kg/día), hipertrigliceridemia (>2.000mg/dl) y proteinuria persistente (3.910mg/24h). El estudio renal (ecografía y biopsia) no detectó ninguna nefropatía estructural. En ese momento se añadió al tratamiento metformina (1.700mg/día), pioglitazona (7,5mg/24h) para mejora de la resistencia a la insulina y enalapril (10mg/día) y losartán (50mg/día) para control de la nefropatía diabética. Desde entonces persistió mal control glucémico (HbA1C 13%) pese al aumento de la cantidad total de insulina diaria hasta 4,2 U/kg/día), hipertrigliceridemia (>2500mg/dl) con desarrollo de esteatosis hepática y deterioro progresivo de la función renal (FG 58ml/min/m2). Hasta el momento el examen de fondo de ojo ha sido normal. Durante la evolución mejoró puntualmente el control glucémico tras terapia con bomba de infusión subcutánea contínua de insulina pero la aparición de abscesos en zonas de inserción del catéter obligó a su retirada.
Actualmente se mantiene con pauta de insulina con bolus preprandiales de insulina aspartica (4,5 U/kg/día). El análisis de los niveles de leptina ha revelado valores indetectables de la misma. Se realizó estudio genético (Hospital Saint Antoine de París) mediante técnica de secuenciación de fragmentos de ADN obtenidos por PCR para búsqueda sistemática de mutaciones constitucionales de la secuencia codificante de los genes de AGPAT2 y BSCL2, donde no se hallaron alteraciones. Una búsqueda posterior por esta misma técnica de alteraciones en el gen LMNA determinó mutación sin sentido en heterocigosis (C.29C>T, I10T) localizada en el exón 1 del gen LMNA que afecta a los dominios N-terminal y C-terminal de las lamininas A y C. Dicha mutación da lugar a una proteína truncada. Esta mutación ya ha sido descrita previamente6,7.
DiscusionPresentamos dos pacientes con síndrome de lipodistrofia congénita generalizada o síndrome de Berardinelli-Seip y diabetes mellitus asociada de difícil control con desarrollo precoz de complicaciones microvasculares. Varios genes han sido implicados en el desarrollo de LCG. Garg et al8 encontraron relación de la LCG con el locus 9q34 (BSCL1). Posteriormente Agarwal et al9 identificaron el gen AGPAT2, que podría causar la LCG inhibiendo la síntesis de tri-acil-glicerol en el adipocito. Magré et al10 identificaron otro locus (BSCL2 o seipina) en el cromosoma 11q13. Se han encontrado mutaciones en el gen de la seipina en familias del Líbano, Turquía, India, Sudáfrica, Brasil y Europa. Finalmente estudios recientes han podido demostrar implicaciones de los genes CAV111,12, PTRF13 y LMNA6,7. El paciente del caso 1, que cumplía todos los criterios mayores para el diagnóstico de LCG (lipoatrofia que afecta al tronco, muslos y cara; rasgos acromegaloides, hepatomegalia, hipertrigliceridemia y resistencia a la insulina), no presentó mutación asociada a estos genes. El paciente del caso número 2 presentaba una mutación puntual en heterocigosis del gen de la laminina. Mutaciones en este gen, se han descrito asociadas a síndromes de lipodistrofia parcial como la progeria de Hutchinson-Gilford o la lipodistrofia parcial familiar tipo Dunningan14-16. Únicamente dos publicaciones relacionan alteraciones en este gen con el desarrollo de lipodistrofia congénita de Berardinelli-Seip6,7. Consideramos que la paciente del caso 2 sería la tercera descripción en la literatura de LCG asociada a mutación en el gen LMNA.
Los individuos afectos de LCG desarrollan resistencia a la insulina asociada al déficit de leptina y aproximadamente un 25-35% desarrollan diabetes mellitus entre los 15-20 años de edad5. La paciente del caso 2 desarrolló diabetes a una edad considerablemente más precoz que los casos descritos hasta ahora para mutaciones de este gen. A menudo se trata de diabetes con difícil control, requiriendo en muchos casos cantidades muy elevadas de insulina. No existen pautas específicas para este tipo de pacientes, por lo cual se suelen emplear regímenes terapéuticos similares a los de pacientes con diabetes tipo 1 y, posteriormente ir aumentando progresivamente las dosis según la resistencia a la insulina mostrada.
Un prometedor tratamiento para los pacientes con LCG lo constituye el empleo de leptina recombinante. En este sentido, varios ensayos clínicos controlados han mostrado beneficios metabólicos tras el empleo de leptina recombinante17-19. Beltrand et al17 demostraron que el tratamiento con leptina recombinante durante 4 meses en un grupo de 9 pacientes con LCG sin diabetes mellitus consiguió reducir las cifras de triglicéridos séricos en un 63%, reducir el volumen del hígado un 20% y mejorar significativamente los parámetros de sensibilidad a la insulina. Oral et al18 demostraron que la terapia con leptina recombinante en pacientes LCG y diabetes mellitus mejoró significativamente el control glucémico, la resistencia a la insulina y la hipertrigliceridemia. No obstante, la disponibilidad de la leptina fuera de ensayos clínicos controlados parece ser limitada y no mejora el aspecto físico de los pacientes20.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Al Dr Olivier Lascols (Hospital Saint-Antoine, París) por la realización de los estudios genéticos de ambos pacientes.
