



Las discinesias paroxísticas son trastornos del movimiento caracterizadas por episodios repentinos de movimientos involuntarios. Se dividen en cinesigénicas, no cinesigénicas e inducidas por el ejercicio. Los autores enfatizan la importancia de la historia clínica y de la descripción de los episodios en el diagnóstico diferencial.
Adolescente de 12 años con episodios de torsión de la lengua y posturas distónicas de las extremidades superiores desencadenados por movimientos súbitos como empezar a correr o bajar escaleras y al comienzo del ejercicio, cesando espontáneamente segundos después. Algunos episodios eran desencadenados por el estrés. En la historia familiar, el padre, tío paterno y hermana también presentaban episodios paroxísticos. El examen neurológico intercrítico fue normal. Se identificó una mutación en el gen PRRT2 asociado con trastornos neurológicos como discinesia paroxística cinesigénica, convulsiones infantiles con coreoatetosis, migraña, ataxia episódica, tortícolis paroxística y retraso cognitivo. El tratamiento con carbamacepina fue eficaz.
Paroxysmal dyskinesias are movement disorders characterized by sudden episodes of involuntary movements. They are divided into kinesigenic, non-kinesigenic, and exercise-induced dyskinesias. Emphasis is made on the importance of the clinical history and fully describing the episodes in the differential diagnosis.
The case is presented of a twelve year-old female with paroxysmal episodes of tongue torsion and dystonic postures of the upper limbs when start running or descending stairs and in the beginning of physical exercise, which ceased spontaneously seconds later. Some episodes were triggered by stress. In family history her father, paternal uncle, and sister also had paroxysmal movements. Interictal neurological examination was normal. Laboratory tests revealed a mutation in PRRT2 gene, which is related to neurological disorders such as paroxysmal kinesigenic dyskinesia, infantile convulsions and choreoathetosis migraine, episodic ataxia, paroxysmal torticollis, and intellectual disability. Treatment with carbamazepine was effective.
Las discinesias paroxísticas son un grupo de trastornos del movimiento caracterizado por episodios repentinos de movimientos involuntarios unilaterales o bilaterales, de tipo coreico, distónico, balístico o mixto. Se dividen en cinesigénicas (DPC), no cinesigénicas (DPNC) e inducidas por el ejercicio (DPE). Son provocados por un factor precipitante, que en la DPC es el movimiento o el cambio repentino de posición; en las DPNC los episodios pueden ser espontáneos o asociados con la fatiga y las crisis en la DPE son desencadenadas por el ejercicio prolongado. En la DPC la mayoría de los casos son de transmisión autosómica dominante, por mutación en el gen de la proteína transmembrana rica en prolina 2 proline-rich transmembrane protein 2 (PRRT2), pero también pueden ser esporádicos o secundarios a infecciones, traumatismos y tóxicos1–5. Los episodios tienen una duración de segundos a minutos y la frecuencia es variable; los niños son los más afectados y los episodios suelen empezar en la primera década de la vida. Los pacientes pueden referir un aura precedente a las crisis, sin pérdida de conciencia durante las mismas. La DPC responde bien a la mayoría de los antiepilépticos en dosis bajas3.
Caso clínicoPaciente de 12 años, sexo femenino, raza negra, fue remitida a la consulta de neuropediatría por episodios paroxísticos de torsión de la lengua y posturas distónicas bilaterales de las extremidades superiores, sin pérdida de conciencia, con un año de evolución. Los episodios ocurrían tras movimientos de inicio repentino, como empezar a correr o bajar escaleras y al comienzo del ejercicio físico. Algunos eran desencadenados por el estrés, siendo interpretados como síntomas de ansiedad. La frecuencia era variable ya que episodios múltiples podrían ocurrir en el mismo día con duración de pocos segundos y sin síntomas postictales. La joven negó experimentar auras o síntomas sensitivos. Y tampoco era capaz de interrumpir la crisis.
La exploración, incluyendo el examen neurológico interictal, fue normal. En las consultas no se observaron crisis.
En los antecedentes familiares (fig. 1): padre y tío paterno con episodios repentinos de torsión del cuello y tronco con predominio por la mañana, al despertar, nunca investigados, y su hermana mayor, de 22 años, con episodios repentinos de dificultad para caminar, «quedaba atrapada» (sic). Los antecedentes personales eran irrelevantes: embarazo normal, parto a término, sin incidentes en el período perinatal; evolución de talla, peso y perímetro cefálico normal y desarrollo psicomotor adecuado. Sin antecedentes de convulsiones, migraña, trauma o infección del sistema nervioso central.
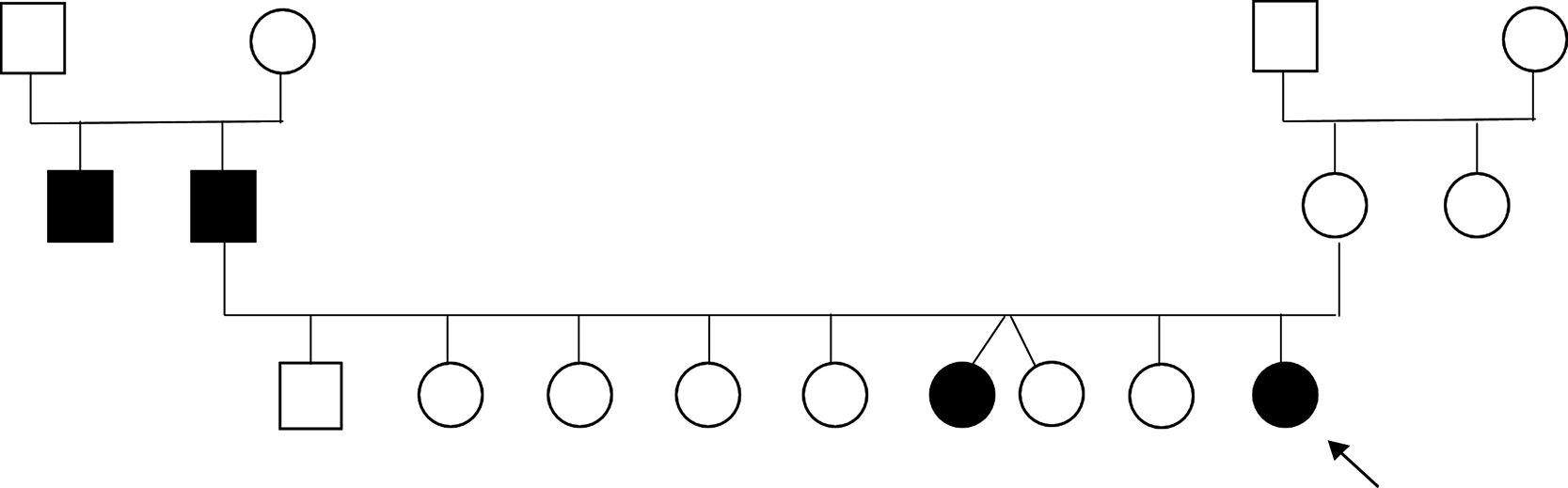
Las pruebas de laboratorio realizadas incluyendo ionograma completo, amonio y función tiroidea fueron normales. Examen citoquímico del LCR y relación glucorraquia/glucemia normales. El electroencefalograma no evidenció actividad paroxística. Por las características de los episodios y patrón familiar fue colocada la hipótesis de DPC y comenzó tratamiento con carbamacepina, 400mg/día (7mg/kg), sin recurrencia de los episodios. Se solicitó el estudio genético, con identificación de la mutación c.649dupG, en heterocigotía, en el gen PRRT2 confirmando la sospecha diagnóstica. Los miembros de la familia sintomáticos fueron remitidos a una consulta de neurología.
DiscusiónEl diagnóstico diferencial de los trastornos del movimiento puede ser muy difícil. Las discinesias paroxísticas son perturbaciones del movimiento raras caracterizadas por movimientos episódicos, con examen neurológico normal entre ellos.
La primera descripción de las discinesias paroxísticas aparece en 1940 cuando Mount y Reback6 describieron una discinesia episódica idiopática e introdujeron el término «coreoatetosis paroxística familiar». Desde entonces, otros casos han aparecido en la literatura y en 1967 Kertesz7 describió casos en que los ataques fueron provocados por movimientos bruscos surgiendo el término «coreoatetosis paroxística cinesigénica». En 1977 Lance8 describe otro tipo de discinesia en que los episodios se desencadenan durante el ejercicio prolongado y que se clasifican como «distonía paroxística inducida por el ejercicio». La clasificación más ampliamente utilizada es la de Dermirkirian y Jankovic9 y que divide las discinesias paroxísticas de acuerdo con los factores desencadenantes y la respuesta al tratamiento en: DPC, DPNC, DPE y discinesias hipnogénicas que se producen durante el sueño, aunque este último se reconoce ahora como una forma de epilepsia3.
La DPC es posiblemente la forma más común, aunque su prevalencia es desconocida10. En 2004 Bruno et al.11 propusieron criterios para identificar las DPC: identificación de los factores precipitantes de la crisis, breve duración (menos de un minuto), sin pérdida de conciencia y sin dolor durante los episodios, la exclusión de otras entidades que puedan justificar los síntomas, el examen neurológico normal entre los episodios, buena respuesta con antiepilépticos.
La fisiopatología de la DPC todavía no está clara. En 2011, Chen et al.4 identificaron 3 mutaciones en el gen PRRT2 en 8 familias con esta enfermedad. Desde entonces, se identificaron otras mutaciones en este gen asociadas con la DPC. El gen PRRT2 se encuentra en el brazo largo del cromosoma 16 y su función no está completamente caracterizada, pero puede estar implicado en la liberación de neurotransmisores en la membrana presináptica11. Mutaciones en el gen PRRT2 son más frecuentes en los casos de transmisión familiar que en los casos esporádicos, y la presencia de mutación en este gen parece asociarse con inicio más temprano de la enfermedad y síntomas no discinéticos. Existe una gran variabilidad fenotípica de mutaciones heterocigotas en el gen PRRT2. La DPC fue el primer fenotipo asociado a esa mutación, pero desde entonces se han identificado también casos de convulsiones infantiles con coreoatetosis (ICCA), epilepsia benigna infantil familiar (BFIE), migraña, ataxia episódica, tortícolis paroxística y convulsiones febriles. Sin embargo, la prevalencia de mutaciones en el gen PRRT2 difiere en los diversos fenotipos, siendo más frecuente en las DPC, ICCA e BFIE que en casos de migraña, ataxia episódica y tortícolis paroxístico. Los diferentes fenotipos pueden ocurrir por separado o en combinación4,5,12–16. Mutaciones homocigotas se asociaron con fenotipos neurológicos más graves con retraso cognitivo, lo que sugiere un efecto aditivo de una dosis doble de la mutación del gen15. La correlación entre las diferentes mutaciones y el fenotipo neurológico todavía no está clara.
El diagnóstico diferencial debe de hacerse con otras enfermedades que pueden cursar con trastornos del movimiento, como las enfermedades neurodegenerativas, metabólicas y endocrinas, por su clínica y alteraciones en las pruebas de laboratorio y de imagen3.
Dentro de las discinesias paroxísticas las características de los episodios y factores desencadenantes son la clave para el diagnóstico diferencial y clasificación: en las DPNC los episodios se desencadenan por otros factores como el consumo de ciertos alimentos como el café o el té, por lo general aparece en la infancia y afecta más a los niños; los pacientes pueden tener episodios múltiples en el mismo día, con duración más larga, 10 a 15min o continuar durante horas. Los episodios son más difíciles de tratar con fármacos antiepilépticos. En las DPE las crisis se desencadenan después del ejercicio continuo superior a 10-15min y la distonía afecta solo los miembros que están en movimiento, con una duración de hasta 60min. Las mutaciones en el gen SLC2A1, responsables por el déficit cerebral del transportador de glucosa tipo 1 (GLUT1) se identificaron en pacientes con la DPE. En estos casos, la relación glucorraquia/glucemia se reduce1.
La carbamacepina es uno de los fármacos más utilizados en la DPC, con una excelente respuesta, pero hay también descripciones de respuestas positivas con varios otros fármacos antiepilépticos 9,17,18.
Se presenta una familia con 4 miembros con DPC, con diagnóstico genético confirmado en el caso índice. Los episodios son muy sugerentes, aunque la clínica sea heterogénea. La investigación etiológica fue negativa hasta la identificación de la mutación en el gen PRRT2. La descripción de los episodios sintomáticos y la forma hereditaria de la transmisión de la DPC hace de éste un diagnóstico probable en los restantes miembros de la familia.
A pesar de tratarse de una enfermedad rara su diagnóstico es crítico, en la mayoría de los casos hay una buena respuesta a la terapia anticonvulsiva. Una descripción exhaustiva de las crisis es vital ya que la investigación etiológica no es informativa. La ausencia de antecedentes familiares y examen neurológico normal pueden retrasar el diagnóstico o incluso dar lugar al diagnóstico erróneo de trastorno psicógeno con retraso en la institución de terapia eficaz.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
El caso clínico descrito se ha presentado previamente en el 14° Congreso Nacional de Pediatría, Porto, Portugal.
