


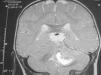
Se comenta el caso de un lactante con un trastorno paroxístico desde la semana de vida en el que se identificó una tumoración de fosa posterior a los 9 meses, 5 meses tras una primera resonancia magnética (RM) normal.
Varón con episodios desde los 7 días de vida de agitación, movimientos continuos de extremidades, facies de sufrimiento, taquicardia, taquipnea, máculas eritematosas en la cara, cutis marmorata en los miembros inferiores y desaturaciones en relación con las apneas. Mantenía el seguimiento visual y no aparentaba alteración de la consciencia. Los episodios, de escasos segundos a 2h de duración, ocurrían 2-4 días a la semana, varias veces a lo largo del día. Los episodios impresionaban por su gravedad aparente, con sensación de riesgo vital inminente y de sufrimiento y desazón extremos del niño.
Antecedentes familiares y personales, sin interés. Desarrollo psicomotor y exploración neurológica normales. Estudios normales: bioquímica, amonio, láctico, aminoácidos, homocisteína, cobre, ceruloplasmina, ácidos grasos de cadena larga, creatincinasa, prueba de transferrina deficiente en carbohidratos, prueba de toluidina, hormonas tiroideas, marcadores tumorales (alfafetoproteína y enolasa neuronal específica), gonadotropina coriónica humana beta, ácidos orgánicos y catecolaminas en orina e investigación de tóxicos, serologías frente a toxoplasma, rubéola, herpes y citomegalovirus, parásitos en heces, EEG intercríticos y críticos, pH-metría, tránsito gastroduodenal, ecografía abdominal, estudio cardiológico y RM cerebral a los 3 meses y medio de edad. No se encontraron mutaciones en el gen SCN9A.
Las crisis no respondieron a diazepam, fenitoína ni valproato por vía intravenosa ni a valproato oral. Con carbamazepina por vía oral los episodios disminuyeron en frecuencia y duración, pero a los 9 meses se añadió un tortícolis indoloro. Una nueva RM cerebral, 5 meses después de la inicial (que revisada a posteriori no muestra ninguna alteración) y tras 1 mes de tortícolis persistente, mostró una tumoración de la fosa posterior (fig. 1). A los 11 meses se realiza una resección subtotal de meduloblastoma con posterior quimioterapia sistémica e intraventricular.
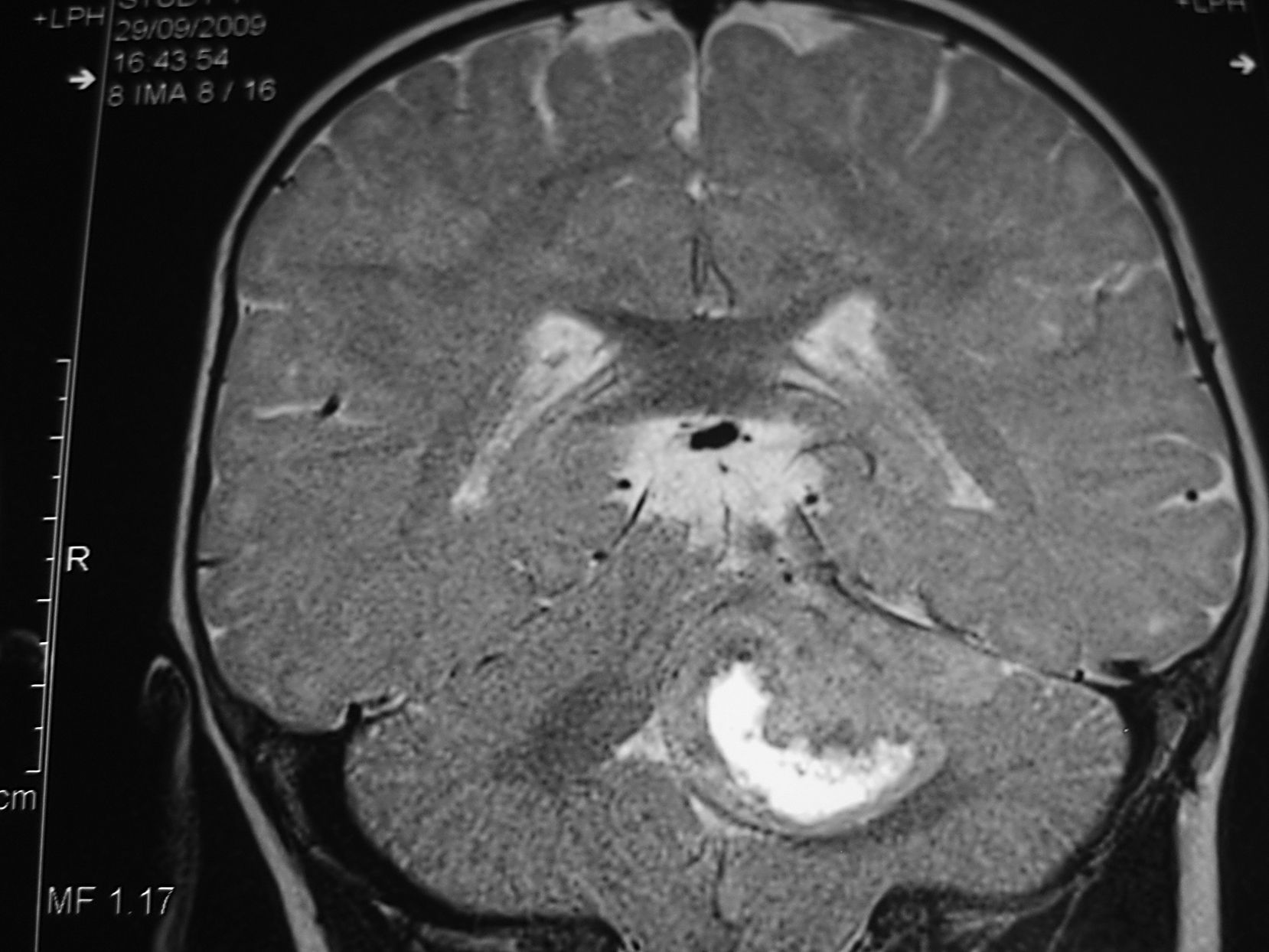
Tras la intervención no volvió a presentar trastornos paroxísticos y a los 3 meses se retiró el tratamiento con carbamazepina. A los 17 meses finalizó el tratamiento quimioterápico, no habiendo signos de recidiva. A los 20 meses tras hidrocefalia aguda que precisó derivación ventriculoperitoneal, se evidenció en la RM diseminación leptomeníngea; no reaparecieron los trastornos paroxísticos. El niño falleció a los 23 meses de edad tras tratamiento quimioterápico paliativo.
Se plantearon diferentes diagnósticos: epilepsia, síndrome de Sandifer, intoxicaciones, tumores secretores de aminas vasoactivas (neuroblastoma, feocromocitoma o síndrome carcinoide, capaces de producir clínica vegetativa), discinesias paroxísticas, taquiarritmias y 2 canalopatías: la hiperekplexia o enfermedad del sobresalto1 y el trastorno de dolor extremo paroxístico2.
El trastorno de dolor extremo paroxístico es una entidad infrecuente (77 pacientes en 5 familias), con herencia autosómica dominante. Es una canalopatía del sodio con mutaciones en el gen SCN9A. Puede comenzar en el periodo neonatal y persiste a lo largo de la vida. Predominan las manifestaciones autonómicas con exantema eritematoso fugaz, coloración en arlequín y movimientos tónicos, pudiéndose llegar a producir síncopes. Los afectados describen los ataques como un dolor punzante e insoportable en la zona rectal, ocular o en áreas de la mandíbula; aunque también puede ser un dolor difuso. Existen diversos factores desencadenantes, como defecación, viento frío, ingesta y emociones. La carbamazepina es parcialmente efectiva en casi todos los casos2.
El tortícolis podría relacionarse con la canalopatía del sodio, dado se han descrito trastornos en los canales iónicos en el tortícolis paroxístico benigno3.
La tumoración de fosa posterior planteó diagnósticos no considerados previamente:
- –
Epilepsia cerebelosa: en niños con astrocitoma de bajo grado o tumoraciones de células displásicas (ganglioglioma, hamartoma y ganglioneurocitoma) que afectan al cerebelo o suelo del cuarto ventrículo. Las crisis aparecen durante el primer año, frecuentemente el primer día de vida, y pueden presentarse hasta 250 veces al día; consisten en repetidas y estereotipadas muecas faciales, desviación de cabeza y ojos, parpadeo, colocación de los miembros en diferentes posturas y manifestaciones vegetativas. Desarrollo psicomotor generalmente normal. Los EEG son normales y habitualmente no hay respuesta al tratamiento antiepiléptico, resolviéndose las crisis sólo tras la resección completa del tumor4,5.
- –
Disfunción autonómica paroxística6: episodios de agitación, sudoración, taquicardia, taquipnea, hipertermia, pudiéndose asociar con posturas de extensión. Se ven en casos de lesiones cerebrales graves, principalmente tras traumatismo craneoencefálico o lesiones hipóxicas y en relación con tumores y quistes supraselares, hidrocefalias o hemorragias intracraneales7,8. Es una entidad pobremente comprendida y no hay consenso en la nomenclatura; existen hasta 31 términos diferentes, siendo los más frecuentemente utilizados: «crisis diencefálicas», «tormentas simpáticas paroxísticas» e «inestabilidad autonómica paroxística con o sin distonía»9. Se ha descrito algún caso de hiperactividad simpática unilateral en relación con siringomielia y/o malformación de Chiari tipo I10,11 y en la anomalía de Chiari tipo II12.
La sintomatología de las crisis de nuestro paciente es compatible con disfunción autonómica, situación poco referida en la literatura en relación con tumores de fosa posterior: no hemos encontrado ninguna descripción en tumores de cerebelo, y sólo una en tumor de tronco (glioma en niño de 7 años)6, que es a su vez el caso descrito más precoz de disfunción autonómica paroxística de origen tumoral. La rareza del caso se incrementa porque no se identificó la tumoración en la RM hecha a los 3 meses de edad y tras 3 meses de sintomatología clínica. Las crisis debían obedecer a células tumorales que ya estaban en periodo neonatal aunque no existía la tumoración identificable por RM.
Diferentes trastornos paroxísticos, incluidas epilepsias, pueden ser clínicamente indistinguibles independientemente de su origen estructural cerebral o no, como es el caso de las canalopatías. Nuestro caso, sin duda excepcional, muestra que la normalidad de la neuroimagen no excluye totalmente el origen estructural, por lo que, en función de la evolución, puede ser necesaria la repetición de la RM cerebral.
