



La fibrodisplasia progresiva osificante (FOP) (MIM:1351000) es una rara enfermedad que afecta al tejido conectivo. Su prevalencia mundial es de 1:2.000.000 habitantes1. Aunque su transmisión es autosómica dominante, en la mayoría de los pacientes se debe a una nueva mutación en familias no afectadas previamente2. Se caracteriza por la formación de hueso heterotópico en tejidos blandos, asociándose a diferentes malformaciones óseas, siendo la más característica la del primer dedo de ambos pies (hallux valgus) que se encuentra presente en el 95%3 de los casos. Generalmente, aparece en edades tempranas, surgiendo los primeros focos de osificación en la primera década de la vida y siendo difícil su diagnóstico en las etapas precoces de la enfermedad. La aparición de los focos de osificación no sigue una regla fija, aunque su distribución suele comenzar de forma craneocaudal y de proximal a distal, siendo las ubicaciones más frecuentes la cabeza, el cuello y la espalda. Esta patología cursa en brotes, pudiendo estos ser desencadenados por traumatismos banales, procedimientos invasivos, infecciones o aparecer de forma espontánea.
La fisiopatología de esta enfermedad no está totalmente aclarada, aunque se ha descrito una sobreexposición de la proteína formadora de hueso (BMP) que podría actuar de señal para la formación de hueso heterotópico4.
La alteración genética de esta patología se debe a mutaciones en el gen ACVR1 localizado en el cromosoma 2q23-24, presentando la mayoría de los pacientes la mutación p.Arg206His5 en heterocigosis. Actualmente están apareciendo publicaciones que describen nuevas mutaciones en el gen ACRV1 asociadas a FOP6.
En la actualidad, ningún tratamiento ha demostrado detener la enfermedad.
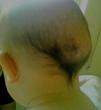
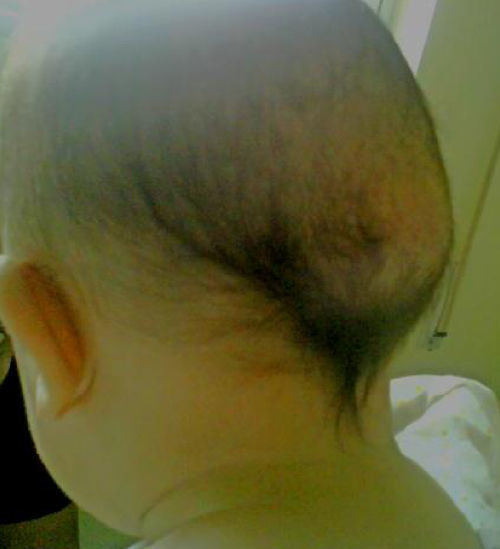
Se presenta a un lactante de 6 meses, ingresado en nuestro hospital por infección del tracto urinario, sin antecedentes familiares ni personales de interés. En la exploración física destacaban diferentes malformaciones esqueléticas como hallux valgus bilateral (fig. 1) y clinodactilia del primer dedo de ambas manos, que según la madre estaban presentes desde el nacimiento. Llamaban la atención 2 tumoraciones subcutáneas a nivel craneal (parieto-temporal izquierda y parieto-occipital derecha), de consistencia pétrea y de aproximadamente 3-4cm de diámetro (fig. 2). Estas habían aparecido a los pocos meses de vida, sin relación con ningún evento traumático. El resto de la exploración fue normal. El hemograma, la bioquímica y análisis del metabolismo fosfo-cálcico fueron normales. Las radiografías de pies y manos realizadas pusieron de manifiesto la malformación ósea del primer dedo de ambos pies y un acortamiento del primer metacarpo. Se realizó una TC craneal que fue normal y no mostraba focos de osificación heterotópica. Ante la sospecha de FOP se solicita estudio genético, el cual mostró en heterocigosis la mutación p.Arg206His (c.617G>A) del exón 7 del gen ACVR1, lo que confirmó el diagnóstico. El paciente completó el tratamiento para la infección urinaria por la que había sido ingresado y fue dado de alta. En la actualidad sigue revisiones periódicas en nuestra unidad, no habiendo presentado hasta el momento ninguna exacerbación de la enfermedad.

La rareza de esta enfermedad hace que la mayoría de las veces pase desapercibida o sea mal diagnosticada, a pesar de que las malformaciones típicas estén presentes desde el nacimiento. Un estudio mostró que hasta en el 87% de los casos publicados el diagnóstico inicial no fue correcto7. Este hecho adquiere especial importancia a la hora de evitar procedimientos invasivos que desencadenen nuevos brotes de la enfermedad y/o tratamientos innecesarios.
Aunque no hay ningún tratamiento curativo, diferentes publicaciones sugieren que el uso de antiinflamatorios puede ayudar a disminuir la intensidad de los brotes8. Los bifosfonatos son otra opción terapéutica, pero no deben ser utilizados de forma rutinaria por sus efectos secundarios. Debemos hacer énfasis tanto en el manejo multidisciplinar que ayude mejorar la calidad de vida y la movilidad de estos pacientes, como en las medidas preventivas. En la actualidad se están desarrollando nuevas modalidades de tratamiento basadas en el uso de anticuerpos monoclonales inhibitorios de la BMP, que en un futuro podrían dar solución a esta enfermedad8,9.
En relación con la evolución, debemos decir que aunque casi todos los casos tenían las deformidades congénitas, la aparición del primer brote de osificación suele ocurrir en torno a los 5 años. El 95% de los pacientes presentaban a la edad de 15 años importantes restricciones en la movilidad de los miembros superiores, mientras que la afectación severa de los miembros inferiores no suele ocurrir hasta más de una década después10. La mayoría sufre en las etapas finales una insuficiencia respiratoria restrictiva por la osificación de los músculos del tórax y por las graves deformidades de la columna. Esto, junto a que los brotes pueden ser desencadenados por infecciones virales, hace pensar en el beneficio de la vacunación antigripal en estos pacientes11.
A pesar de ser encontrada en referencias bibliográficas desde la antigüedad, son muchas las sombras que todavía nos quedan por descubrir sobre esta terrible enfermedad, lo cual debe ser un incentivo para seguir trabajado hacia un futuro más esperanzador.
