






Los defectos oculares congénitos (DOC) pueden originar importante discapacidad.
ObjetivoEl objetivo de este estudio fue conocer la prevalencia total de los DOC en Asturias, su tendencia y realizar una descripción de su forma de presentación.
MetodologíaAnálisis de los datos del Registro de Defectos Congénitos de Asturias (RDCA) de los años 1990–2004. La población estudiada fueron los 103.452 nacidos de madres residentes en Asturias en este periodo. Se calcularon las tasas de prevalencia total.
ResultadosDe los 3.035 casos con defectos congénitos registrados durante los 15 años estudiados, 70 tenían un DOC. La prevalencia total media fue de 6,8 por 10.000 nacidos, con una tendencia estable. Los más frecuentes fueron: las cataratas congénitas (2,0 por 10.000 nacidos vivos), la anoftalmia/microftalmia (1,4 por 10.000 nacidos vivos) y los colobomas (1,3 por 10.000 nacidos vivos). El 40 % de los DOC se presentaron de forma aislada, 37% pertenecían a un síndrome y 23% se asociaban a otras anomalías congénitas no sindrómicas.
ConclusionesLa prevalencia total de los DOC durante este periodo en Asturias fue estable siendo las cataratas congénitas el DOC más frecuente. Más de la mitad de los DOC, en especial la anoftalmia/microftalmia se asociaron a otras malformaciones congénitas.
Congenital ocular anomalies (COAs) can produce serious disability.
ObjectiveThe purpose of this investigation was to assess the prevalence of COAs, their trends and to describe the associated malformations and syndromes in a geographically defined population.
MethodData from the Asturias Registry of Congenital Defects were used. The period studied was from 1990 to 2004 and the study population the 103,452 births of mothers living in the region. Total prevalence was calculated.
ResultsA total of 3035 cases with congenital defects were recorded, of these 70 had COAs. The total prevalence was 6.8 per 10000 births, with a stable trend during this period. The most common COAs were: congenital cataract (2.0 per 10000 births), anophthalmos/microphthalmos (1.4 per 10000 births) and coloboma (1.3 per 10000 births). 40% of COAs occurred as isolated defects, 37% were syndromes and 23% were associated with other congenital defects.
ConclusionsThe prevalence of COAs in Asturias over this period had a stable trend and the congenital cataract was the commonest COAs. COAs, particularly the anophthalmos/microphthalmos were associated with other congenital anomalies.
Las anomalías oculares congénitas constituyen un grupo de defectos congénitos (DC) que pueden generar grave discapacidad, puesto que el sentido de la vista es esencial para el aprendizaje, la comunicación y la relación social. Por ello, la detección precoz de los defectos oculares congénitos (DOC) es fundamental: permitirá su corrección total o parcial, así como atención temprana y educación especial para minimizar sus efectos, cuando no sea posible evitar la ceguera parcial o total. Aunque pueden presentarse aisladas, con frecuencia se asocian a otras alteraciones del desarrollo, con afectación de otros órganos o sistemas, como ocurre en los síndromes1.
No existen muchas publicaciones acerca de la epidemiología de los DOC2–4 o bien proceden de grupos seleccionados (déficits visuales5, Down6, retraso mental7, etc.). Por esta razón, hemos realizado este trabajo con el objetivo de conocer la frecuencia de los DOC en nuestro medio, su evolución temporal y forma de presentación.
Material y métodosEl Registro de Defectos Congénitos de Asturias (RDCA) es un registro de base poblacional cuyas características están descritas en su manual operativo y recogidas en la página web del European Concerted Action on Congenital Anomalies and Twins8 (EUROCAT), con el que comparte, en lo fundamental, una metodología común.
La población objeto de estudio fueron todos los nacidos (vivos y muertos) y abortos inducidos (AI) por DC de mujeres residentes en Asturias desde el 1 de enero de 1990 al 31 de diciembre de 2004.
El RDCA realiza un sistema de vigilancia y captación de casos diagnosticados hasta los 5 años de vida. Las fuentes de información utilizadas en esta captación sistemática de casos fueron las de los servicios de Pediatría y Neonatología. Anualmente se realiza una búsqueda activa cruzando la base de datos de los casos ya captados con otras múltiples fuentes de información9. Los casos de DC registrables incluyen los definidos por el EUROCAT y excluye malformaciones menores aisladas.
La codificación de los DC se realizó con la CIE-9 (Clasificación Internacional de Enfermedades) hasta el año 2000 y con la CIE-10 a partir de entonces. Incluimos los siguientes defectos oculares: anoftalmia/microftalmia (A/M) unilateral o bilateral, cataratas, colobomas y otros defectos oculares que incluyen malformaciones congénitas de los párpados, del aparato lagrimal, de la órbita, y del globo ocular. Se excluyen escleróticas azules siguiendo los criterios del EUROCAT.
Se considera que A/M constituyen expresiones o grados diferentes de una misma alteración de desarrollo prenatal, por lo que se suelen estudiar en conjunto1,9.
El número total de casos no se corresponde con la suma de DOC ya que en 4 casos existían 2 defectos oculares combinados, los cuales se tabularon separadamente en cada tipo de defecto.
Para el estudio de las frecuencias de DOC se utilizó como indicador principal la tasa de prevalencia total: total de casos (vivos, muertos y AI) registrados por cada diez mil nacidos (vivos y muertos). Con el fin de obtener tasas más estables, hemos agrupado los casos en cinco periodos de 3 años. Se utilizó el test de chi cuadrado para analizar la homogeneidad de las tasas y la tendencia. Para la comparación con otros registros europeos se calcularon los IC del 95% de las tasas correspondientes. Se tomó una significación estadística del 5%.
ResultadosEn el período estudiado se han registrado 3.035 casos de DC en un total de 103.452 nacidos. El número de casos con DOC fue de 70, con una prevalencia total de 6,8 casos por diez mil nacidos (IC 95%: 5,2–8,4).
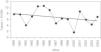
La tabla 1 muestra la prevalencia del conjunto de los DOCs así como de los más frecuentes: catarata congénita, A/M y colobomas. Las prevalencias trianuales fueron homogéneas para el total de DOC (chi cuadrado para homogeneidad 2,93; p=0,56) con tendencia ligeramente descendente no significativa (chi cuadrado 0,88, p=0,34). La figura 1 muestra esta evolución temporal. El pequeño número de casos de los defectos separadamente no permite un análisis de tendencia consistente.
Prevalencia total (tasa por 10.000) del total de los defectos oculares congénitos y de algunos específicos (catarata congénita, anoftalmia/microftalmia y coloboma) en Asturias, 1990–2004
| Totales | Catarata congénita | Anoftalmia/microftalmia | Coloboma | ||||||
| Año | Nacidos | Casos | Prev tot | Casos | Prev tot | Casos | Prev tot | Casos | Prev tot |
| 1990–92 | 22.889 | 15 | 6,6 | 5 | 2,2 | 6 | 2,6 | 3 | 1,3 |
| 1993–95 | 20.191 | 19 | 9,4 | 4 | 2,0 | 5 | 2,5 | 3 | 1,5 |
| 1996–98 | 19.301 | 13 | 6,7 | 8 | 4,1 | 0 | 0,0 | 1 | 0,5 |
| 1999–01 | 20.067 | 11 | 5,5 | 1 | 0,5 | 2 | 1,0 | 3 | 1,5 |
| 2002–04 | 21.004 | 12 | 5,7 | 3 | 1,4 | 1 | 0,5 | 3 | 1,4 |
| 1990–04 | 103.452 | 70 | 6,8 | 21 | 2,0 | 14 | 1,4 | 13 | 1,3 |
Las cataratas congénitas fueron el DOC más frecuente en nuestro estudio. El grupo de otros DOC incluyó: seis casos de ptosis palpebral, uno de blefarofimosis congénita bilateral y uno de dacriocistitis congénita del ojo derecho; 6 con alteraciones del segmento anterior del ojo: anomalía de Axenfeld-Rieger (1), aniridia bilateral (1), heterocromia del iris (3), anomalía de Peters (1); 8 con malformaciones del segmento posterior del ojo: vítreo primario hiperplásico persistente (1), hemorragia vítrea y retiniana (1), alteraciones de la retina (un caso de atrofia, uno de distrofia y otro de desprendimiento), 2 con persistencia de la arteria hialoidea bilateral y uno de retinoblastoma, y otras (un caso con quiste pedunculado del borde externo del ojo y uno con glaucoma congénito bilateral).
La mayoría (67) fueron nacidos vivos; se registró un AI: polimalformado con A/M bilateral asociado a osteocondrodisplasia y adactilia en las 4 extremidades y dos nacidos muertos, uno con A/M presente en el síndrome de Neu Laxova y otro cuyo único hallazgo fue una catarata aislada.
Respecto al sexo del nacido, se encontró un predominio de varones (59,4%), con una razón de masculinidad de 1,37, a expensas del grupo de otros DOC (19 varones y 7 mujeres), puesto que la proporción fue similar entre ambos sexos tanto en los casos con cataratas congénitas (10 varones y 11 mujeres), colobomas (7 varones y 6 mujeres) como en los A/M (8 varones y 5 mujeres). En el AI el sexo fue desconocido.
En relación con el momento del diagnóstico, 50 casos (71,4%) se diagnosticaron al nacer o antes de la primera semana de vida; 11 entre el primer mes y el año de vida: 4 casos de cataratas, 2 de colobomas, uno de blefarofimosis bilateral, uno de glaucoma bilateral congénito, uno con aniridia bilateral, un síndrome de Marcus Gunn y uno de Noonan; de estos 9 (81,8%) eran DOC aislados. Dos se detectaron pasado el primer año de vida, correspondiendo a una ptosis palpebral en un paciente con síndrome de Noonan y a un síndrome de Senior-Loken. En seis el diagnóstico fue prenatal, debido a la presencia de otros defectos asociados o a formar parte de un síndrome: tres A/M con alteraciones del SNC y una trisomía 18; un coloboma con alteración cromosómica estructural (translocación 5;6) y múltiples malformaciones; un síndrome de Down con dacriocistitis congénita; y un caso con hemorragia vítrea y alteraciones del SNC.
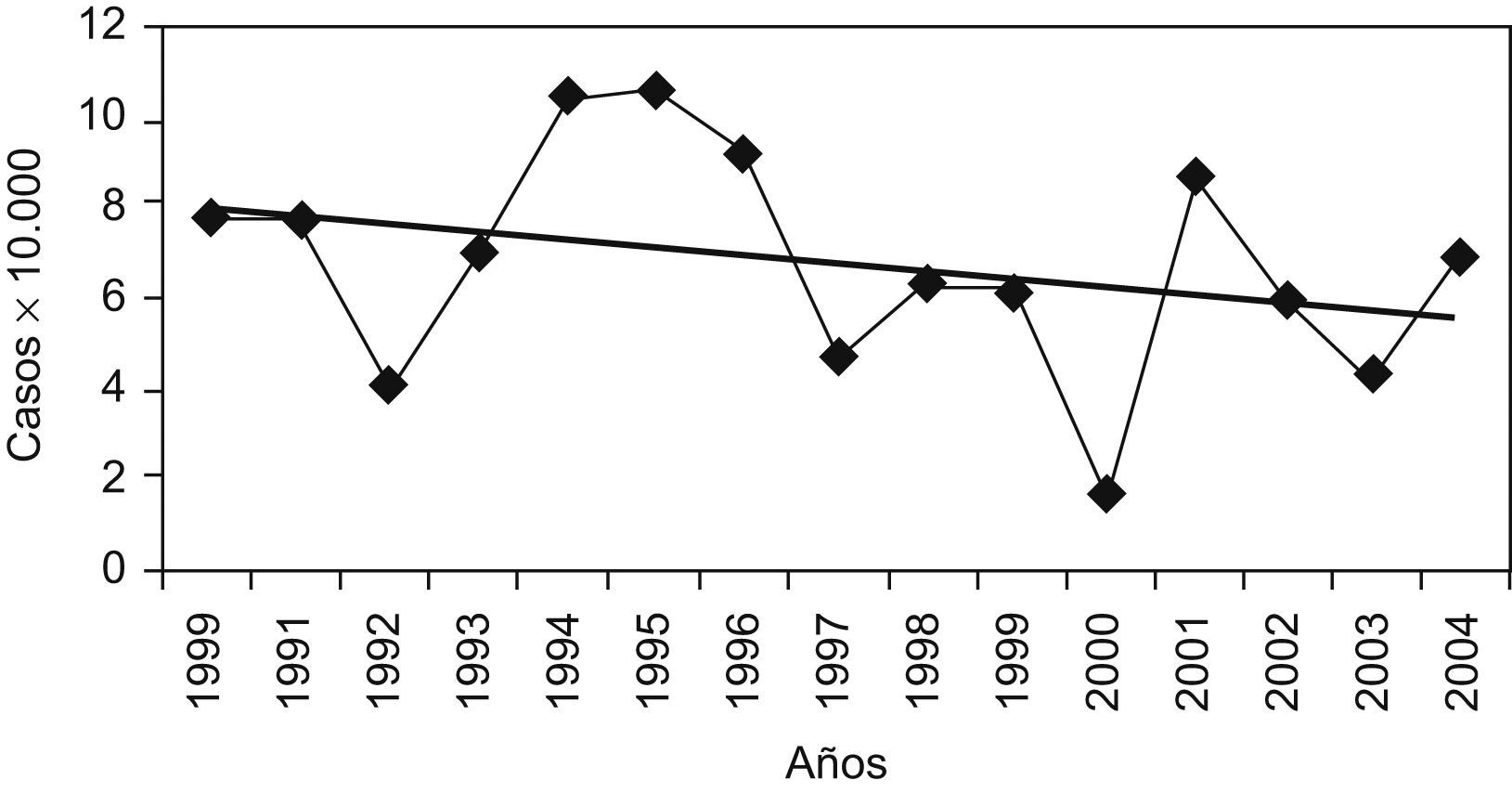
El 60% del total de DOC, tenían otra/s anomalía/s extraoculares asociada/s. Esta frecuencia de asociación difiere según el tipo de DOC (fig. 2), siendo del 38% en las cataratas congénitas, mientras que alcanzó el 79% en las A/M. Las malformaciones asociadas eran muy heterogéneas. En la tabla 3 se detallan los síndromes identificados en los DOC registrados. La cromosomopatía más frecuente con DOC fue el síndrome de Down (3 casos). También hubo tres casos de síndrome de Noonan y otros tres con síndrome de Goldenhar. Entre los polimalformados que no pertenecían a ninguna entidad sindrómica reconocida, las alteraciones del SNC fueron las más frecuentes, seguidas de las fisuras labiales y palatinas. Se incluye el denominado síndrome o fenómeno de Marcus Gunn que a pesar de su nombre no es un síndrome y que en el seguimiento posterior presentó parálisis cerebral.
Cuatro casos presentaron dos DOC combinados: una cromosomopatía (17 q+) y una toxoplasmosis tenían catarata congénita en un ojo y microftalmia en el otro, un caso con síndrome de Morning Glory tenía microftalmia y coloboma papilar y otro presentaba microftalmia y coloboma de iris.
Globalmente, se realizó estudio cromosómico en 40 casos (57,1%), en un porcentaje similar en los distintos tipos de DOC.
En cinco casos de cataratas congénitas aisladas existían antecedentes familiares de dicha patología. El caso de colobomas bilaterales con translocación (5;6) también tenía antecedentes familiares de coloboma. Asimismo, la madre del caso con anomalía de Axenfeld-Rieger tenía glaucoma bilateral y ceguera.
DiscusiónLos DOC supusieron un pequeño número del total de DC registrados en Asturias durante los 15 años estudiados10. Sin embargo, la discapacidad que ocasionan justifica su estudio detallado5.
El RDCA está constituido por un grupo de trabajo estable y cohesionado lo que, probablemente, reduce la variabilidad interpersonal entre los pediatras participantes y mejora la calidad clínica en la descripción de los casos. Además, la metodología seguida, en especial la utilización de múltiples fuentes permitió exhaustividad por lo cual consideramos que nuestros datos presentan gran fiabilidad y son una buena medida de los DC, en particular de los DOC en Asturias. Sin embargo, también tienen algunas limitaciones, como son las deficiencias en el estudio postmortem de los AI, que podrían llevar a un subregistro de algunos DOC especialmente aquellos asociados a cromosomopatías y/o polimalformaciones, al no disponer de una descripción detallada de los mismos. Este hecho podría explicar la menor frecuencia de A/M, DOC frecuentemente asociado con cromosomopatías, a partir del año 1996, coincidiendo con la mejora del diagnóstico prenatal y un aumento de AI por DC en nuestra comunidad autónoma. En este sentido, cabe señalar, por ejemplo, que de los 47 casos de trisomía 18 registrados durante los 15 años del estudio11, tan solo en uno de ellos se ha descrito la existencia de A/M.
Por otra parte, la prevalencia de DOC en el RDCA fue estable en este periodo, ligeramente superior a la media del EUROCAT y a la del País Vasco y similar a la del registro de Barcelona (tabla 2). Las cataratas congénitas fue el DOC más frecuentemente registrado en nuestros datos y su tasa también fue más alta que en los otros registros; estas diferencias, aunque relevantes, no son estadísticamente significativas.
Comparación de las tasas de prevalencia del conjunto de defectos oculares congénitos y de aquellos más frecuentes, en Asturias, el EUROCAT y otros registros españoles de base poblacional
| Asturias 1990–2004 | EUROCAT 1990–2004 | REDCB 1992–2003 | RACAV 1990–2004 | |||||
| N.° casos | Prev Total | N.° casos | Prev Total | N.° casos | Prev Total | N.° casos | Prev Total | |
| Totales | 70 | 6,8 (5,2–8,3) | 4075 | 4,6 (3,3–5,9) | 90 | 5,4 (3,9–6,8) | 102 | 4,0 (2,8–5,2) |
| Catarata congénita | 21 | 2,0 (1,2–2,9) | 904 | 1 (0,4–1,6) | 12 | 0,7 (0,2–1,2) | 30 | 1,2 (0,5–1,8) |
| Anoftalmia/microftalmia | 14 | 1,4 (0,6–2,1) | 1013 | 1,1 (0,5–1,8) | 24 | 1,4 (0,7–2,2) | 28 | 1,1 (0,5–1,7) |
EUROCAT: European Concerted Action on Congenital Anomalies and Twins; Prev Total: tasas por 10.000 nacidos (IC del 95%); RACAV: Registro de anomalías congénitas de la Comunidad Autónoma del País Vasco; REDCB: Registro de Defectos Congénitos de Barcelona.
Fuente de los datos: EUROCAT26, elaboración propia.
Síndromes identificados en los defectos oculares registrados
| Tipo | Cataratas | Micro/anoftalmia | Colobomas | Otros | Total casos¿ |
| Down (T21) | 1 | 2 | 3 | ||
| Patau (T13) | 1 | 1 | |||
| Edwards (T18) | 1 | 1 | |||
| 10 q+ | 1 | 1 | |||
| 17 q+ | 1 | 1 | 1 | ||
| 13 p- | 1 | 1 | |||
| Translocación (5;6) | 1 | 1 | |||
| Noonan | 3 | 3 | |||
| Silver-Russell | 1 | 1 | |||
| Neu-Laxova | 1 | 1 | |||
| Morning Glory | 1 | 1 | 1 | ||
| Lowe | 1 | 1 | |||
| Goldenhar | 1 | 2 | 3 | ||
| Bardet Bield | 1 | 1 | |||
| CHARGE | 1 | 1 | |||
| Waadenburg tipo II | 1 | 1 | |||
| Orstavik | 1 | 1 | |||
| Senior Loken | 1 | 1 | |||
| Toxoplasmosis | 1 | 1 | 1 | ||
| Rubéola | 1 | 1 |
Diversos estudios de búsqueda activa y con múltiples fuentes de información en el Reino Unido muestran que la prevalencia de cataratas congénitas comunicadas en el primer mes de vida difiere de la recogida cuando se realiza un seguimiento a lo largo de la infancia12. Las diferencias al alza del RDCA podrían explicarse por la inclusión de casos hasta los 5 años de vida, pudiendo incluir casos de detección más tardía, mientras que otros registros hacen seguimiento hasta el primer año. No obstante, tan solo 2 DOC fueron de diagnóstico posterior al primer año, por lo cual la mayor frecuencia registrada quizás esté más influida por la búsqueda activa y el empleo de múltiples fuentes de información13. En cambio, nuestras tasas de A/M fueron similares tanto a la media del EUROCAT como a los registros de Barcelona y País Vasco y también coincidente con otros estudios. Así, Busby et al9 encontraron una tasa de A/M del 1,0 y el Registro de Alberta (Canada)14 un 1,2 por 10.000 nacidos. Dado que la A/M con gran frecuencia se asocia a otras malformaciones extraoculares, principalmente cromosomopatías y malformaciones del SNC y musculoesqueléticas es más difícil que pase desapercibida. Aunque no podemos hablar de las tendencias individuales de DOC específicos, parece que en los 3 últimos trienios la frecuencia de A/M fue menor. Como ya se ha comentado podría deberse a un subregistro por no descripción detallada de los DC de los síndromes, en especial de las cromosomopatías.
No hemos comparado nuestras prevalencias con las del Estudio Colaborativo Español de Malformaciones Congénitas (ECEMC) dada la diferente metodología seguida. Sin embargo, como hemos señalado, incluso los registros de base poblacional, más sensibles y con menos sesgos que los de base hospitalaria, podrían estar influenciados por otras diferencias metodológicas (método de búsqueda de los casos, fuentes, criterios de inclusión, límite de edad para el momento del diagnóstico, precisión y método de confirmación del diagnóstico entre otros)15, que justificarían las diferencias encontradas.
La razón de masculinidad en los DOC fue de 1,37 mientras que en el conjunto de DC del RDCA fue 1,33 siendo en el conjunto de nacidos de Asturias de 1,1. El registro húngaro4 también encuentra mayor frecuencia en varones. Por el contrario, Stoll et al3 hallan mayor prevalencia de DOC en mujeres (razón de masculinidad 0,82). Si excluimos los casos de hipospadias, que podrían introducir un sesgo, el índice de masculinidad de los DC en el RDCA sería 1,2, es decir, parece haber un exceso de DC y DOC en el sexo masculino.
Los DOC tienden a presentarse asociados a otras anomalías, siendo imprescindible un estudio detallado para confirmar o descartar la presencia de otros defectos. En nuestro medio tres de cada cinco DOC se asociaban a otras malformaciones. Nuestro porcentaje de detección de casos aislados es superior al de otras series16, posiblemente por la exhaustividad del registro y la inclusión de casos hasta los 5 años. La A/M fue la que más frecuentemente se asoció a otras malformaciones, sindrómicas o no sindrómicas. Hay que destacar la frecuente asociación con conocidas cromosomopatías, concretamente los hemos encontrado en un caso de trisomía 13, una trisomía 18 y 2 trisomías parciales, así como en síndromes poco frecuentes como el Neo Laxova y el de «Morning Glory»17.
Cabe destacar la heterogeneidad etiológica en los casos de DOC. Tal y como queda reflejado en la tabla 3, hemos detectado 20 tipos diferentes de entidades desde cromosomopatías y síndromes génicos a otros síndromes de etiología ambiental como la embriofetopatía rubeólica y la infección por toxoplasma, también presentes en otras series1. Es importante una cuidadosa historia prenatal, buscando infecciones agudas, exposiciones a teratógenos o enfermedades crónicas maternas. Asimismo, se recogerá si existen antecedentes familiares de alteraciones oculares, que fueron positivos en un 10% en nuestro estudio, sobre todo en las cataratas congénitas aisladas lo cual es sugerente de alteración genética, de transmisión autosómica dominante.
Por otro lado, del diagnóstico etiológico depende el asesoramiento a la familia acerca de los riesgos de recurrencia, detección de portadores y posibilidad de diagnóstico prenatal. En nuestros datos, de los 40 casos que contaban con cariotipo, 9 (22,5%) presentaban una cromosomopatía; dada esta alta frecuencia de cromosomopatías asociadas es fundamental la realización sistemática de estudio citogenético, siendo imprescindible en los casos de A/M polimalformados.
El rápido avance científico de la genética molecular permite el diagnóstico de un número cada vez mayor de alteraciones genéticas, no detectables con el estudio cromosómico habitual, posibilitando el conocimiento de la etiopatogenia de algunos DOC. Así se han identificado genes como el SOX218, cuyas mutaciones se asocian con la A/M, y del PAX2 con coloboma, bien aislado o con anomalías urogenitales19, entre otros. Por tanto, es aconsejable recoger muestras adecuadas, también en AI y nacidos muertos que permitan realizar estos estudios.
El síndrome de Down fue la cromosomopatía más frecuente que hemos encontrado en los DOC, aunque también es la más frecuentemente registrada, presentando un caso coloboma de retina, uno dacriocistitis congénita y otro heterocromia del iris. La gran variabilidad de los síntomas oculares en este síndrome20, podría relacionarse con el polimorfismo resultante de las copias extra de los genes presentes en el cromosoma 2121.
Se ha implicado a las técnicas de reproducción asistida con la aparición de DOC22. En nuestro estudio, solamente en un caso que presentaba hemorragia vítrea y malformaciones del SNC se recogió el antecedente de dichas técnicas. El intercambio de información entre los registros de técnicas de reproducción asistida y los de DC permitirán conocer si realmente suponen un riesgo aumentado de DOC.
Las cataratas congénitas fue el DOC más prevalente en nuestro estudio; aunque sólo un caso estuvo ligado a infección rubeólica y otro a infección por toxoplasma en los 15 años de estudio (en los que se registraron dos casos de rubéola congénita y tres de toxoplasmosis), éstos serían susceptibles de prevención primaria (inmunización frente a rubéola, medidas para evitar infección por toxoplasma durante el embarazo entre otras) o bien de prevención secundaria, que se apoya en un diagnóstico y tratamiento precoz.
Todo cuanto interfiera en el proceso de aprendizaje visual del cerebro provocará ambliopía, es decir, reducción de la visión por falta de estimulación visual adecuada durante el periodo crítico de desarrollo visual. Cuanto más temprana y prolongada sea la interferencia con la visión tanto más profunda será la ambliopía. Por tanto, el factor decisivo para el éxito del tratamiento es el diagnóstico temprano, que asegure el tratamiento efectivo y la educación a la familia del niño o niña afectos. Así, la intervención quirúrgica precoz en la persistencia de la arteria hialoidea puede evitar la ambliopía23.
Prácticamente todos los grupos de expertos recomiendan examinar a los recién nacidos y lactantes para el despistaje de alteraciones oculares y defectos de alineación ocular24. La exploración cuidadosa de los ojos en el neonato y el lactante permite detectar alteraciones del tamaño ocular, del tamaño, forma y transparencia de la córnea, presencia de cataratas, luxación del cristalino, aniridia, colobomas y ptosis palpebral entre otros. Se explorará la motilidad ocular, el reflejo fotomotor y la presencia de reflejo retiniano de color rojo. La leucocoria, es decir el reflejo pupilar blanco, es la primera manifestación del retinoblastoma. Nuestro caso con retinoblastoma presentaba deleción del brazo corto del cromosoma 13 (13p−). Una encuesta realizada en el Reino Unido, pone de manifiesto la necesidad de mejorar el examen neonatal rutinario25. Un juguete vistoso que haga ruido para atraer la atención del bebé y una linterna de bolsillo son suficientes para la exploración ocular de neonatos, lactantes y niños de hasta 3 años. Por tanto, en nuestro país con una cobertura sanitaria pública universal, el papel de los pediatras de atención primaria, a través de los controles de salud periódicos va a ser clave para detectar aquellos casos afectos de DOCs que no hayan sido diagnosticados en el hospital. De igual modo, es fundamental el seguimiento, vigilando la aparición de retrasos.
En conclusión, la prevalencia total de los DOC en Asturias durante los 15 años estudiados fue del 6,8 por 10,000 nacidos. Cabe destacar que un alto porcentaje de A/M presentan malformaciones asociadas. Por otro lado, recalcamos la importancia de un examen ocular minucioso en la exploración rutinaria del neonato y del lactante para detectar precozmente estos DC, disminuir o atenuar la frecuente discapacidad que conllevan y mejorar la calidad de vida de los niños y niñas afectos de estos defectos así como de sus familias.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
FinanciaciónConsejería de Salud y Servicios Sanitarios.
Este trabajo ha sido posible gracias a las gerencias y trabajadores de los hospitales públicos del Principado de Asturias (Hospital de Jarrio, Hospital Carmen y Severo Ochoa, Hospital San Agustín, Hospital Universitario Central de Asturias, Hospital de Cabueñes, Hospital Grande Covián, Hospital Valle del Nalón, y Hospital Álvarez Buylla), que han colaborado en la recogida de datos y facilitado en todo momento nuestra labor.
