




La granulomatosis de Wegener pertenece al grupo de vasculitis de vaso pequeño asociadas a anticuerpos anticitoplasma de neutrófilo. Se caracteriza por inflamación granulomatosa y vasculitis necrosante que puede afectar a diversos órganos, principalmente cursa con afectación renal y de las vías respiratorias. Es una enfermedad rara en la infancia y el diagnóstico precoz es fundamental para el pronóstico a largo plazo de la enfermedad. La presencia de anticuerpos anticitoplasma de neutrófilo con patrón citoplasmático o de títulos elevados de anticuerpos antiproteinasa-3 ha sido actualmente incluida en los criterios diagnósticos de vasculitis en niños. En este artículo se presenta un caso de granulomatosis de Wegener en la infancia, con presencia de anticuerpos anticitoplasma de neutrófilo con patrón citoplasmático con ausencia de anticuerpos antiproteinasa-3 y presencia de niveles elevados de anticuerpos anticatepsina G, raramente asociados a granulomatosis de Wegener.
Wegener's granulomatosis belongs to the group of small vessel vasculitis associated with anti-neutrophil cytoplasmic antibodies characterized by granulomatous inflammation and necrotising vasculitis in various organs with particular involvement of the upper and lower respiratory tracts and kidneys. Wegener's granulomatosis is a rare disorder in childhood and early diagnosis of this disease is critical to the long-term prognosis of the disease. The presence of positive cytoplasmic antineutrophil cytoplasmic antibody staining or a high titre of proteinase 3 antibodies were added as new criteria of vasculitis in childhood. This article presents a case of Wegener's granulomatosis, with the presence of anti-neutrophil cytoplasm antibodies with cytoplasmic pattern with absence of anti-proteinase 3 antibodies and presence of high levels of anti-cathepsin G antibodies, rarely described in Wegener's granulomatosis.
La granulomatosis de Wegener (GW), actualmente denominada poliangeítis granulomatosa, pertenece al grupo de vasculitis de vaso pequeño asociadas a anticuerpos anticitoplasma de neutrófilo (ANCA). Se caracteriza por la inflamación y la necrosis de los vasos sanguíneos. Es una enfermedad rara en la infancia y el diagnóstico diferencial suele ser complicado, ya que sus manifestaciones clínicas son inespecíficas y heterogéneas. Las diferentes características etiológicas, clínicas y pronósticas de esta enfermedad en niños1,2 han llevado a la definición de nuevos criterios diagnósticos3, entre los que se incluyen, a diferencia de los establecidos en población adulta4, la presencia de ANCA con patrón citoplasmático (C-ANCA) en presencia o ausencia de anticuerpos antiproteinasa-3 (PR3). La metodología tradicionalmente empleada para la determinación de ANCA incluye la inmunofluorescencia indirecta (IFI) sobre neutrófilos fijados en etanol y el enzimoinmunoanálisis para la cuantificación de los antígenos PR3 y mieloperoxidasa (MPO)5. Mediante IFI se identifican fundamentalmente 2 patrones, citoplasmático (C-ANCA) y perinuclear (P-ANCA), que generalmente se corresponde con los antígenos PR3 y MPO, respectivamente. Pero también se ha descrito la presencia de otros antígenos menores, como catepsina G, lactoferrina, proteína de incremento de la permeabilidad bacteriana, elastasa y lisozima, que se asocian a otras afecciones pero que raramente también han sido descritos en vasculitis6.
Presentamos uno de los pocos casos referidos en España de GW en la infancia que, por su escasa incidencia y por la presencia inusual de anticuerpos anticatepsina G asociados a vasculitis, nos parece relevante comunicar.
Caso clínicoNiño de 11 años de edad, sin antecedentes personales ni familiares de interés, que un mes antes de su ingreso comenzó con cuadro catarral y fiebre de 2-3 días de evolución que cedió sin antibioterapia. A la semana presentó artritis en el codo derecho, con inflamación, eritema y dolor que remitieron con antiinflamatorios, reapareciendo en las semanas siguientes en articulaciones alternantes, con carácter migratorio. Durante estas semanas presentó febrícula, con temperatura máxima de 37,8°C hasta 24 h antes del ingreso, cuando reinició cuadro febril de 38,8°C y aumento de los dolores articulares de forma generalizada y con artritis evidente en el tobillo derecho, que motivó el ingreso hospitalario para estudio.
La exploración física al ingreso mostró palidez, astenia y ojeras, con aftas orales de 3 días de evolución. No distrés respiratorio, tonos rítmicos sin soplos aparentes, abdomen blando discretamente doloroso a la palpación en el epigastrio. Lesión macular eritematosa en la rodilla derecha y en el tórax. Afectación articular del codo izquierdo e inflamación de tobillo. En las pruebas complementarias destacaron hemoglobina 8,3g/dl, ligero descenso de proteínas totales 6,3g/dl y aumento de proteína C reactiva 110,4mg/l. El análisis urinario mostró indicios de proteínas (no cuantificadas).
La evolución del paciente fue irregular, con empeoramiento del estado general, fiebre, descenso de hemoglobina hasta 7,7g/dl, dolores articulares de carácter migratorio que no respondieron a tratamiento con analgesia y dolor abdominal. El paciente ingresó en la UCI pediátrica para transfusión de concentrado de hematíes y monitorización. A los 2 días, tras estabilización, regresó a planta.
A los 3 días comenzó con tos productiva, expectoración de moco con sangre roja y dolor costal generalizado, con desaturación basal del 88%. Sangre en evacuaciones y en vómitos. Se realizó una tomografía computarizada de tórax que mostró opacidades parenquimatosas de morfología nodular bilateral, con patrón en vidrio deslustrado, y derrame pleural compatible con vasculitis.
El paciente presentó insuficiencia renal progresiva, con aumento de cifras de urea y creatinina hasta 80mg/dl y 1,8mg/dl, respectivamente. El análisis de orina reveló la existencia de proteinuria (1.192mg/24h) y microhematuria, por lo que el paciente es trasladado a la UCI pediátrica.
Se solicitó un estudio inmunológico, en el que se detectaron ANCA mediante IFI sobre neutrófilos fijados con etanol y formalina, con patrón citoplasmático y título superior a 1/1280 (C-ANCA positivo) (fig. 1). No se detectaron anticuerpos A-MPO, A-PR3 y antimembrana basal glomerular. En el estudio mediante enzimoinmunoanálisis (ELISA) de otras especificidades antigénicas asociadas a ANCA (lactoferrina, catepsina G, proteína inhibidora de la permeabilidad, elastasa y lisozima) se encontraron niveles elevados de anticuerpos anticatepsina G (> 100U/ml).
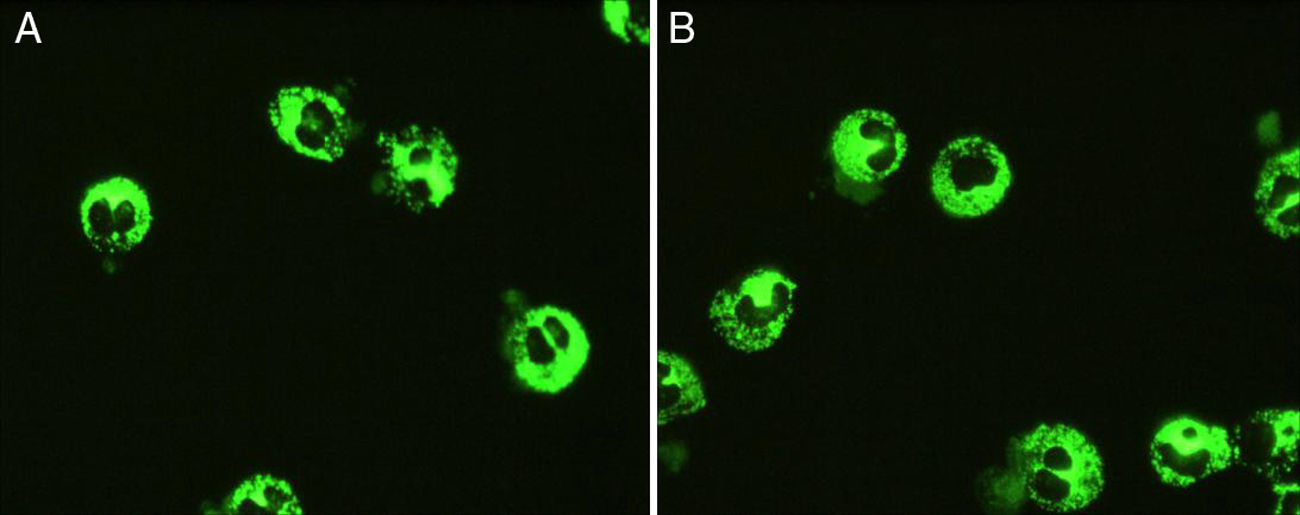
Para la confirmación del diagnóstico de vasculitis, se realizó una biopsia renal en la que se observaron lesiones compatibles con glomerulonefritis extracapilar pauciinmune, con semilunas epiteliales en glomérulos y vasculitis activa (fig. 2).
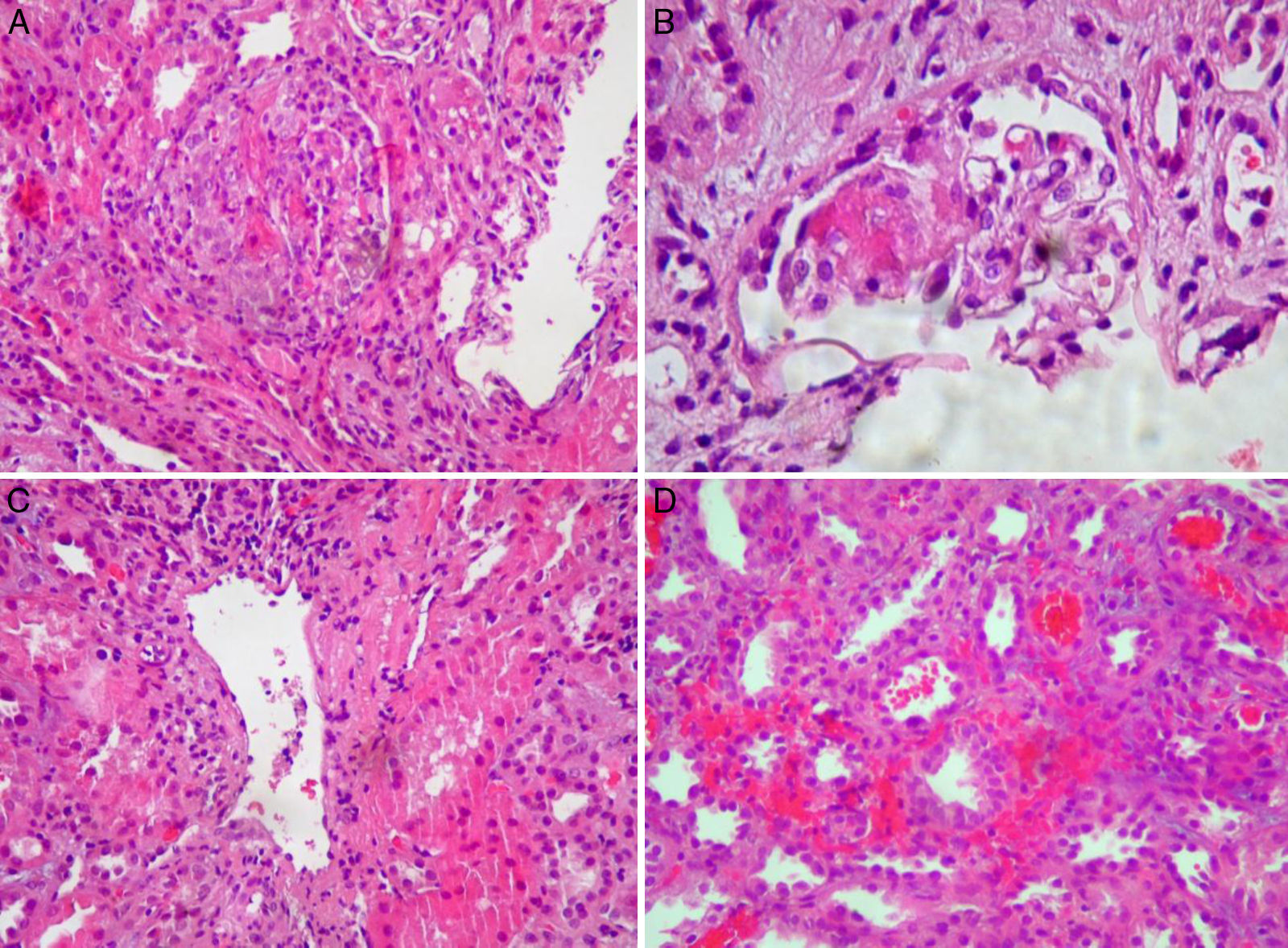
A) Glomérulo mostrando marcada proliferación extracapilar epitelial, con necrosis fibrinoide central. Hematoxilina-eosina, 10×. B) Detalle de penacho glomerular con mínima proliferación extracapilar y necrosis segmentaria. Hematoxilina-eosina, 40×. C) Presencia de vasculitis en vaso de tipo venular. Hematoxilina-eosina, 10×. D) Áreas de extravasación hemática intersticial y hemorragia intratubular.
Se inició tratamiento combinado con corticoides, ciclofosfamida y plasmaféresis, con buena respuesta al tratamiento. Fue dado de alta con mantenimiento de prednisona de 45mg/día. Aunque actualmente se encuentra asintomático, en los controles mensuales posteriores al diagnóstico persiste la presencia de C-ANCA y anticuerpos anticatepsina G a títulos similares a los del inicio. Será necesario la determinación de los niveles de anticuerpos anticatepsina G en controles posteriores para valorar la evolución de los mismos en función de los signos clínicos del paciente.
DiscusiónEl paciente es diagnosticado de vasculitis sistémica, compatible con GW de acuerdo con los nuevos criterios clínicos de vasculitis3. El paciente cumplía 4 de los 6 criterios diagnósticos definidos para GW; microhematuria y proteinuria, inflamación granulomatosa en biopsia renal, afectación de las vías respiratorias y presencia de C-ANCA a título elevados (el paciente no recibió medicación previa que pudiera inducir la presencia de C-ANCA). La vasculitis sistémica es una afección rara en la infancia, aunque se han descrito casos de púrpura de Schönlein-Henoch, síndrome de Churg-Strauss y GW en la infancia, este último como situación clínica excepcional7,8. La presentación clínica de la GW en los primeros estadios de la enfermedad es inespecífica, lo que hace que el diagnóstico diferencial sea complicado. Los dolores articulares, las lesiones cutáneas, la fiebre y las alteraciones del estado general fueron las primeras manifestaciones clínicas, que precedieron en un mes a los síntomas respiratorios en forma de hemorragia pulmonar, hemorragias digestivas y afectación renal, que orientaron al diagnóstico final de GW en este paciente. Estas manifestaciones clínicas, aunque inespecíficas, coinciden con las descritas por Belostotsky y Kostianovsky en otras series de pacientes pediátricos diagnosticados de vasculitis sistémica9,10.
Las diferentes características clínicas y pronósticas de las vasculitis en niños, lleva en 2006 a definir nuevos criterios diagnósticos de vasculitis, y que se introduce la presencia de C-ANCA3. La determinación de ANCA es una prueba no invasiva para el paciente y que es fundamental para la orientación diagnóstica y para el seguimiento de las vasculitis. En pacientes con vasculitis sistémicas primarias, tales como la GW, poliangeítis microscópica y el síndrome de Churg-Strauss, los patrones de tinción IFI corresponden, en general, con anticuerpos frente a los 2 antígenos principales: el patrón C-ANCA se asocia generalmente a anticuerpos frente a PR3 y el patrón P-ANCA con anticuerpos frente a MPO. Sin embargo, algunos sueros positivos para ANCA por IFI son negativos para anti-PR3 y anticuerpos anti-MPO, lo que sugiere la presencia de anticuerpos frente a antígenos menores de los gránulos de los neutrófilos. Entre los antígenos menores se encuentran la elastasa, la lactoferrina, la catepsina G, la lisozima y la proteína inhibidora de la permeabilidad. Estos anticuerpos dirigidos frente a antígenos menores están asociados a otras enfermedades pero también se han descrito raramente en vasculitis. En una serie de 12 pacientes diagnosticados de GW11 se detectaron anticuerpos C-ANCA en 6 pacientes, y en un caso se encontraron anticuerpos anticatepsina G en ausencia de A-PR3 y A-MPO. En 5 casos no se detectaron ANCA por inmunofluorescencia aunque si se detectaron anticuerpos A-MPO y A-PR3, lo cual pone de manifiesto que ante una elevada sospecha clínica de vasculitis y ante un resultado negativo de ANCA por inmunofluorescencia es preciso analizar la presencia de A-MPO, A-PR3 y otros antígenos menores, como ponen de manifiesto las últimas recomendaciones EULAR5.
La determinación de ANCA, actualmente incluidos en los criterios diagnósticos de vasculitis, es fundamental para el diagnostico de GW. La presencia de C-ANCA, incluso en ausencia de A-PR3, puede ser un criterio diagnóstico de GW. En este caso, resulta especialmente interesante la presencia de anticuerpos anticatepsina G en GW, aunque el papel de estos autoanticuerpos en vasculitis sistémica está aún por determinar.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
