



El hiperinsulinismo congénito rara vez aparece asociado a cuadros sindrómicos, y en estos casos suele presentar buena respuesta al tratamiento con diazóxido1.
El síndrome de hipercrecimiento más frecuentemente asociado con hipoglucemia por hiperinsulinismo es el síndrome de Beckwith-Wiedemann (SBW), pero solo un 5% presentan hipoglucemias persistentes más allá del periodo neonatal2. En el síndrome de Sotos (SSo.) (hipercrecimiento pre y posnatal, macrocefalia, edad ósea acelerada y retraso mental), la hipoglucemia por hiperinsulinismo persistente es mucho menos frecuente. Se ha descrito un solapamiento fenotípico entre ambos síndromes, demostrándose en algún caso dicho solapamiento a nivel molecular3.
Presentamos una paciente hija única de padres no consanguíneos. Fruto de gestación espontánea, antecedentes familiares sin interés, nace con 36 semanas de edad gestacional por desprendimiento de placenta. Al nacimiento presenta Apgar 8/9, peso en -0,89DE, longitud en+1,75DE, sin datos del perímetro cefálico. Con 48 horas de vida ingresada por ictericia en su hospital de referencia presenta hipoglucemias no cetósicas (glucemias entre 25-45mg/dl), insulinemia máxima registrada: 7μU/ml coincidiendo con glucemia de 33mg/dl. Requirió glucosa intravenosa a más de 6mg/kg/min. Se completa estudio de hipoglucemia (IGF-1, GH, cortisol, amonio, láctico, carnitina, aminoácidos, ácidos grasos en sangre y ácidos orgánicos en orina) siendo normal. Por persistencia de hipoglucemias con 32 días de edad posnatal se inicia tratamiento con diazóxido e hidroclorotiazida (a 15mg/kg/día y 5,5mg/kg/día, respectivamente), pudiendo suspender la glucosa intravenosa un día después.
A los 2 meses de vida es vista en nuestro servicio destacando en la exploración: hipertricosis, macrosomia, rasgos toscos, frente abombada, hipertelorismo, surcos transversales en hélix y hernia umbilical. Se sospecha un SBW siendo descartado genéticamente. Se completa el estudio genético detectándose una mutación compatible con SSo. en el intrón 14 del gen NSD1 que lleva a alteración del splicing [c.5190+2A (IVS14+2G>A)].
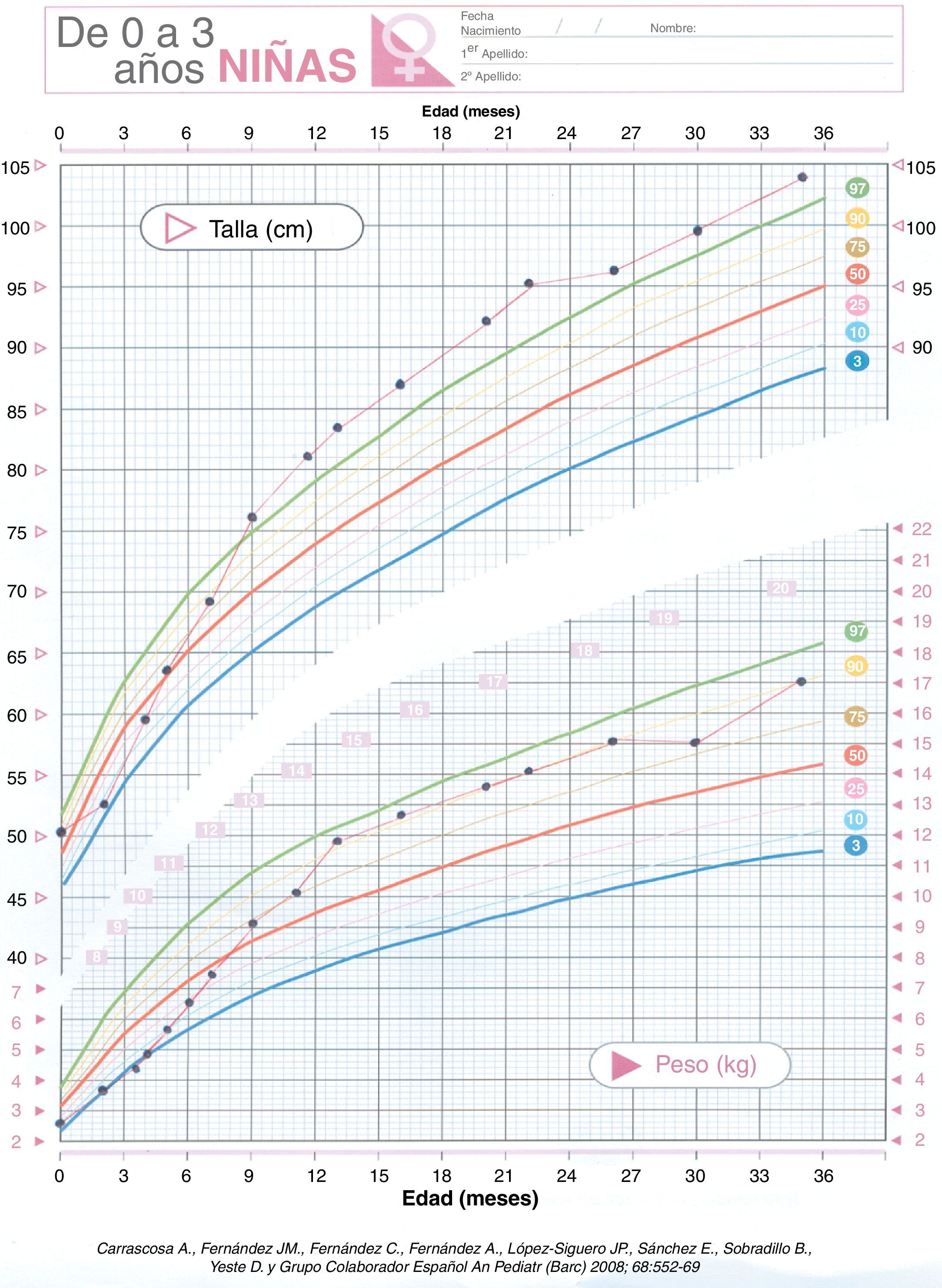
Se realiza ecografía abdominal (riñón izquierdo hipoplásico), alfa-fetoproteína, βHCG, catecolaminas en orina y estudio cardiológico que son normales. La edad ósea está acelerada. En su evolución presenta crisis febriles (EEG y RMN normal), hiperactividad y moderado retraso psicomotor. En cuanto al control glucémico se puede ir disminuyendo la dosis de fármacos lentamente ya que ante mínimos descensos presenta hipoglucemias. Suspendiéndose la hidroclorotiazida con 16 meses de edad. En la actualidad con 4 años realiza dieta acorde a su edad y mantiene glucemias normales con mínima dosis de diazóxido (0,5mg/kg/día). La curva ponderoestatural (fig. 1) ha sido ascendente (peso:+1,78DE, talla:+3 DE y perímetro cefálico:+1,7 DE)
En 1964 Juan Sotos describe este síndrome, treinta años después Cole y Hughes sugieren unos criterios diagnósticos4. Es en el año 2002 cuando Kurotaki et al describen la haploinsuficiencia del gen NSD1 como principal causa del SSo5.
Es probablemente el segundo síndrome de hipercrecimiento más frecuente tras el SBW6 (prevalencia de 1/14.000 recién nacidos vivos).
Los hallazgos más constantes son hipercrecimiento pre y posnatal, rasgos físicos característicos y problemas de aprendizaje7. La altura tiende a normalizarse tras la pubertad pero la macrocefalia persiste. La edad ósea está acelerada (76% de casos4).
Los rasgos faciales más característicos son: frente prominente, escaso pelo frontoparietal, fisuras palpebrales antimongoloides, rostro alargado, mentón puntiagudo, hipertelorismo y dolicocefalia.
Al nacimiento es frecuente encontrar ictericia (65%), hipotonía (5%) y problemas alimentarios (70%). Otros hallazgos son cardiopatía (20%), escoliosis (30%), nefropatía (15%) y crisis convulsivas (25%). Menos frecuentes son las alteraciones oftalmológicas, otitis y criptorquidia. La hipoglucemia con hiperinsulinismo persistente es excepcional.
Por otro lado, se conoce la existencia de una relación entre hipercrecimiento y tumores. Concretamente en el SSo. la incidencia de neoplasias es del 2 a 7%, la mayoría hematológicas a diferencia del SBW donde destacan los tumores abdominales8. Es fundamental un seguimiento mediante exploración física, ecografías abdominales y marcadores tumorales.
Ante sospecha de SSo. debe realizarse estudio genético. La edad ósea (acelerada) y RM cerebral (dilatación ventricular) apoyan el diagnóstico.
La haploinsuficiencia del gen NSD1 (Nuclear Receptor Set-domain-containing protein), en el brazo largo del cromosoma 5 (5q35.3) es responsable de al menos el 75% de SSo5.
Este caso nos parece interesante por existencia de hipoglucemia hiperinsulínica persistente, hallazgo excepcional en el SSo. y por solapamiento clínico con el SBW (hernia umbilical y fisuras en hélix).
Como en nuestra paciente ya ha sido descrito solapamiento fenotípico entre SBW y SSo. En 2004, Baujat et al3 describen mutaciones del NSD1 en dos casos de SBW y, paradójicamente, anomalías en la región 11p15 (típica del SBW) en dos pacientes con diagnóstico clínico de SSo.
Nuestro caso reabre el debate sobre el papel que la proteína NSD1 jugaría en mantener la impronta genética de la región 11p153, explicando el solapamiento clínico entre ambos síndromes.
