
Recién nacido a término, varón, sin antecedentes familiares de interés, que nace mediante parto eutócico con Apgar 9-10. Es el segundo hijo de padres no consanguíneos. No abortos previos ni alteraciones en los controles del embarazo.
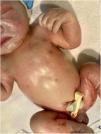
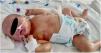
A la exploración destacan piel seca y eritematosa, con descamación y lesiones petequiales generalizadas (figs. 1 y 2) junto con aspecto hipotrófico, ictericia y succión débil.
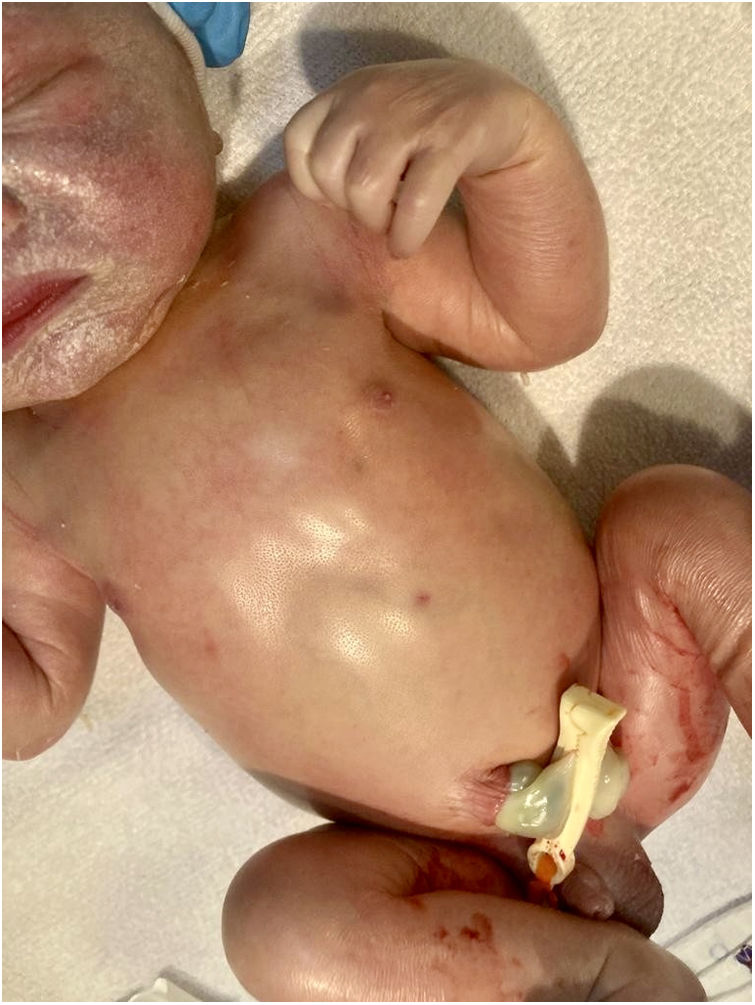
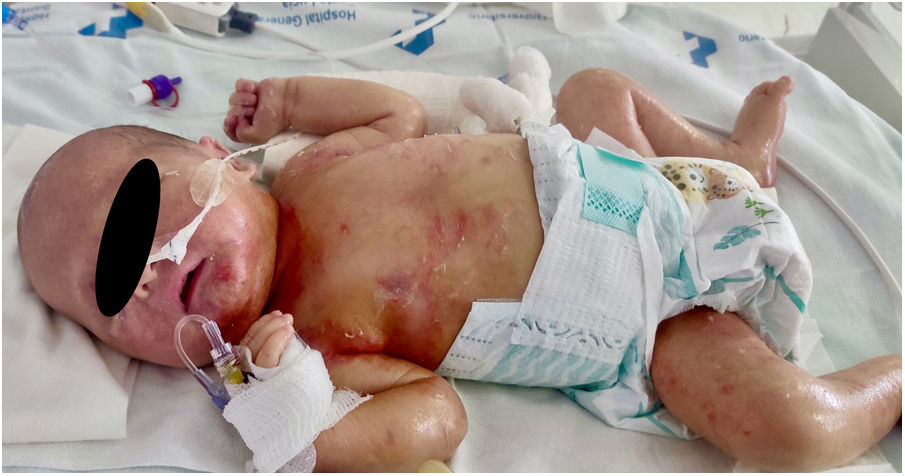
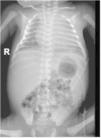
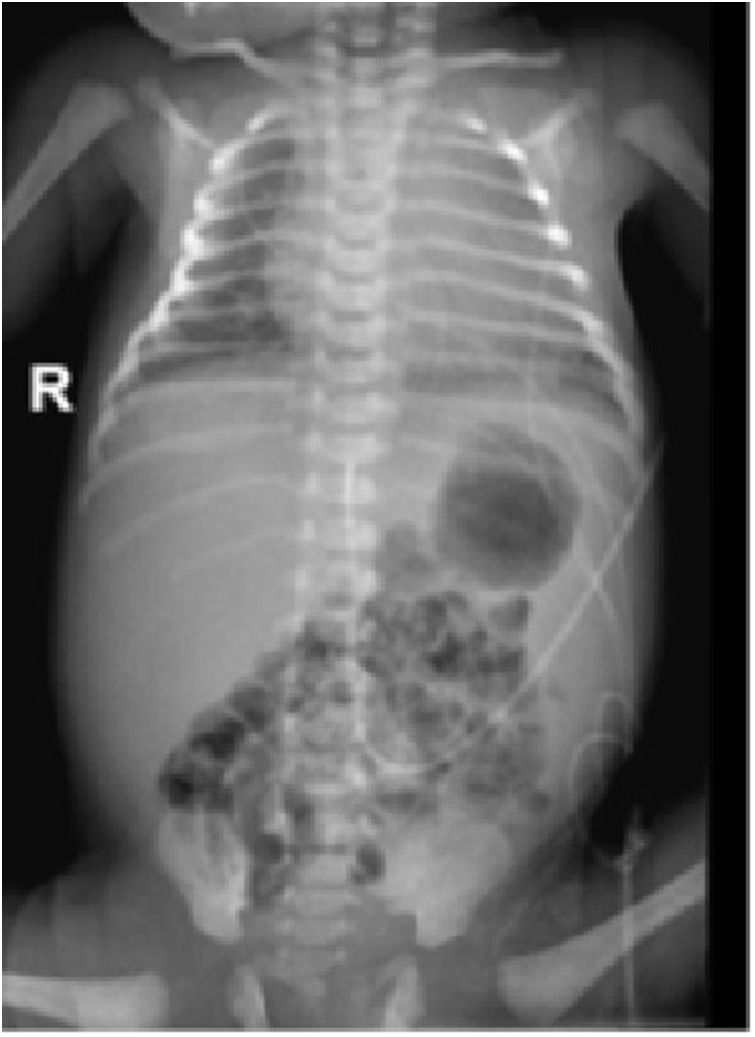
Ingresó en la unidad de cuidados intensivos, evidenciándose coagulopatía, colestasis y trombocitopenia mantenida a pesar de múltiples transfusiones. Los cultivos y serologías fueron negativos. En el frotis de sangre se evidenciaron linfocitos vacuolados. La radiografía y la ecografía posnatal mostraron hepatoesplenomegalia (fig. 3).
Se sospechó ictiosis sindrómica, incluyendo la enfermedad de Gaucher fetal, el síndrome de Chanarin-Dorfman y el síndrome NISCH. El estudio enzimático para enfermedad de Gaucher solicitado al octavo día de vida mostró ausencia de la enzima glucocerebrosidasa. Posteriormente se pidió un panel genético de ictiosis, y se añadió estudio de enfermedad de Gaucher y NISCH. El estudio genético detectó dos mutaciones heterocigóticas del gen GBA (p.Pro430Leu y p.Leu483Pro, presentes en los progenitores). El paciente falleció 2 meses después en el contexto de una infección respiratoria.
La enfermedad de Gaucher fetal1 presenta una incidencia <1/106 en los recién nacidos vivos, y una prevalencia de casi 0 debido a la ausencia de tratamiento efectivo, mortalidad temprana y presentación como abortos o hydrops. En el resto de casos se asocia a hepatoesplenomegalia, citopenias mantenidas, y puede presentarse como un síndrome ictiosiforme2.
Está causada por mutaciones en el gen GBA (1q21), provocando déficit de glucocerebrosidasa. Si asocian la variante p.Leu483Pro, presentan peor pronóstico3.
Es importante conocer la relación entre la trombocitopenia y la ictiosis para poder diagnosticar a estos pacientes con el objetivo de brindar consejo genético a las familias.
