



El síndrome de Peutz-Jeghers (SPJ) es una enfermedad hereditaria rara caracterizada por máculas pigmentadas en la piel y la mucosa oral, y pólipos gastrointestinales múltiples de tipo hamartomatoso. La localización más frecuente de las lesiones polipoideas es el yeyuno e íleon pero también pueden presentarse en estómago, duodeno y colon. Es poco frecuente en la infancia y la clínica de presentación suele ser como invaginación o sangrado intestinal.
Presentamos el caso de un niño con SPJ que debutó como una invaginación gastroduodenal, localización inusual ya que la mayoría de las invaginaciones son intestinales y solo hay descritos 5 casos en la literatura.
Varón de 7 años, que acude al hospital por dolor abdominal, vómitos no biliosos y estreñimiento de varios días de evolución. El paciente presenta historia previa de dolor abdominal recurrente y anemia ferropénica crónica. En los antecedentes familiares destaca la presencia de poliposis colónica en el padre. En la exploración física se observa palidez cutáneo-mucosa, lentiginosis en la mucosa oral y labial, y abdomen con distensión epigástrica.
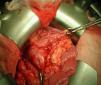
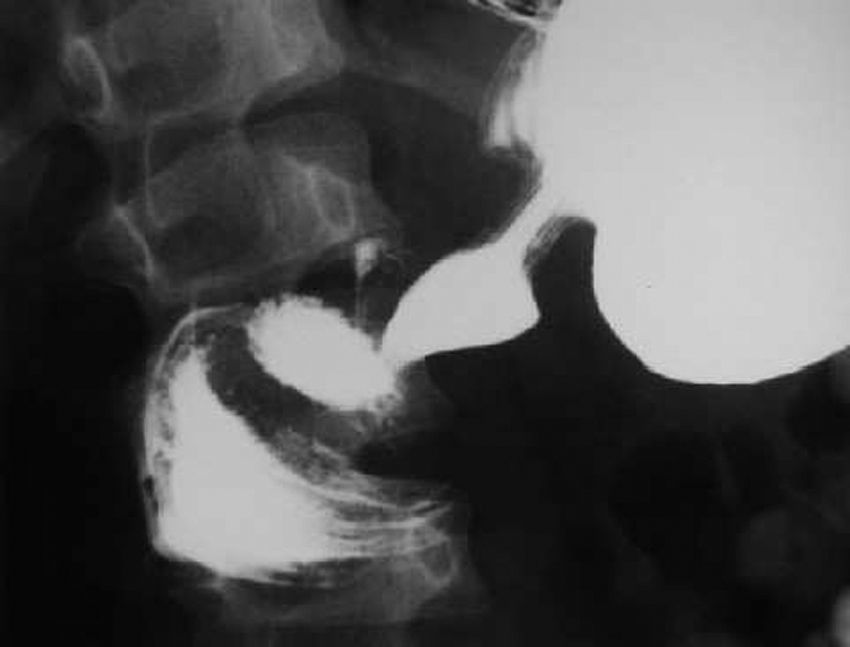
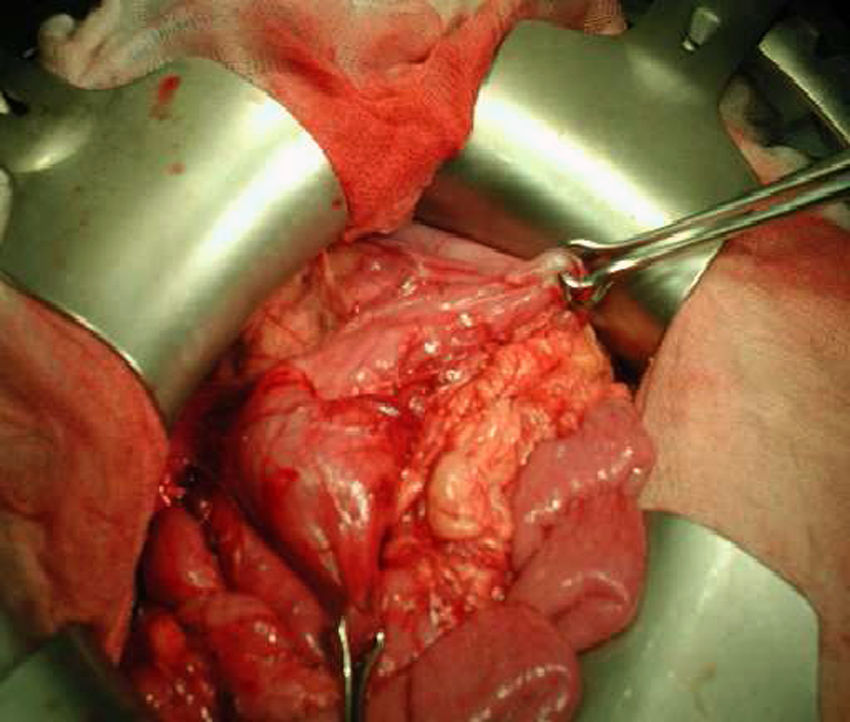
La analítica solo muestra elevación moderada de amilasa 292 U/l (19-161 U/l). Se realiza tránsito intestinal con contraste en el que se observa obstrucción a la salida del estómago con acortamiento antral y defecto de llenado de la segunda porción duodenal (fig. 1). La laparotomía confirma la invaginación gastroduodenal (fig. 2) y la presencia de dos pólipos sésiles de 3 x 5cm y 4 x 2cm, realizándose una desinvaginación manual y gastrostomía antral con exéresis de las lesiones polipoideas. El estudio anatomopatológico es compatible con pólipos de tipo hamartomatoso. La recuperación del paciente es satisfactoria y a lo largo de su seguimiento se han observado numerosos pólipos en el estómago, el duodeno, el colon y el sigma, sin que haya presentado nuevas complicaciones.
La prevalencia estimada del SPJ es de 1 cada 8.300 a 1 cada 280.000 personas1. Se transmite de forma autosómica dominante, con una penetrancia variable, aunque en el 25% de los pacientes se produce por mutaciones de novo2. Está causado por mutaciones en el gen STK11 (también llamado LKB1) localizado en el cromosoma 19p13.3. El gen STK11 codifica una serina-treonina cinasa que actúa como supresor de tumores3. Las mutaciones de este gen se detectan en más del 50% de los individuos con SPJ y su ausencia en familias con diagnóstico clínico de SPJ sugiere que puede haber otro locus genético causante del SPJ4.
El diagnóstico se realiza por la presencia de pólipos hamartomatosos confirmados histológicamente y al menos dos de los siguientes criterios clínicos: historia familiar, hiperpigmentación y pólipos en el intestino delgado5.
Histológicamente, se caracterizan por poseer un eje conectivo fino y muy ramificado que contiene músculo liso y les confiere un aspecto arborescente.
Las máculas pigmentadas están presentes en el 95% de los pacientes5 y son más frecuentes en labios, mucosa bucal y área periorbitaria, aunque también pueden aparecer en los dedos, palmas, plantas y mucosa perianal e intestinal. La mediana de edad de comienzo de los síntomas es a los 13 años y los más frecuentes son dolor abdominal, invaginación y obstrucción del intestino delgado, sangrado rectal y anemia. Presentan múltiples pólipos hamartomatosos que se localizan preferentemente en el intestino delgado, aunque también pueden estar situados en estómago, colon y recto6, y son causa de dolor abdominal recurrente a causa de invaginaciones. También se han descrito pólipos en otras localizaciones, como nasofaringe, vesícula biliar y vejiga urinaria.
El tratamiento de los pólipos obstructivos debe ser endoscópico siempre que sea posible, o mínima resección intestinal para reducir el riesgo de síndrome de intestino corto y otras complicaciones.
El SPJ predispone a cáncer, fundamentalmente en la edad adulta, de origen tanto gastrointestinal como extraintestinal2. El riesgo de cáncer a lo largo de la vida varía entre el 37 y el 93% en comparación a la población general7.
En los niños con SPJ el cribado endoscópico mediante colonoscopia se recomienda cada 3 años comenzando cuando aparecen los síntomas o en la adolescencia temprana si no hay síntomas. A partir de los 10 años se recomienda endoscopia alta bianual y exploración radiológica con bario del aparato digestivo superior2.
En la literatura hay numerosos casos descritos de invaginación intestinal en el SPJ pero se han descrito solo 5 casos de obstrucción a la salida del estómago debido a invaginación gastroduodenal por pólipos gástricos en el SPJ, solo uno de ellos en la infancia6,8-11. El diagnóstico diferencial debe realizarse con otras causas de obstrucción a la salida del estómago, como enfermedad ulcerosa péptica, vólvulo, ingestión de cáusticos, estenosis pilórica, tumores o seudoquistes pancreáticos.
Aunque se trata de un síndrome poco frecuente, puede manifestarse en la infancia, por lo que debe tenerse en cuenta en el diagnóstico diferencial de pacientes con historia familiar positiva que presenten síntomas obstructivos o sangrado intestinal.
